Rare macular manifestation of Alport syndrome. A case report
doi: 10.55342/szemhungarica.2025.162.1.38
Case report
Summary
Alport syndrome is the hereditary disorder of type IV collagen of the basal membrane, characterized by progressive renal impairment, sensorineural hearing loss, cornea, lens, and retina disorders. Frequent manifestations in the eye are recurrent cornea erosion, anterior lenticonus, central and peripheral dot and fleck retinopathy.
Case Presentation: A 38-years-old male patient visited our clinic due to blurred vision for 2 months. He had previously undergone renal transplantation as a consequence of chronic glomerulonephritis and suffered from hypertension. Upon ophthalmic examination, besides the normal anterior segment, no lenticonus was discovered; the mid-peripheral retina did not show signs of dot and fleck retinopathy; however, we found pigment irregularities in the macula. A macula OCT scan was performed, which revealed macular atrophy. Fluorescein angiography showed perimacular and macular hyperfluorescent dots, without leakage. Later the patient visited an otorhinolaryngology clinic due to bilateral tinnitus and hearing impairment; the performed examinations proved bilateral sensorineural hearing loss
Conclusion: Along with the frequently seen corneal erosions, anterior lenticonus, and dot and fleck retinopathy, macular atrophy can occur as a rare ocular manifestation in Alport syndrome. Awareness about this condition is crucial in predicting the prognosis of the vision.
Összefoglaló
Az Alport-szindróma a bazálmembrán IV. típusú kollagénjének örökletes zavara, amely progresszív vesekárosodást, szenzorineurális hallásromlást, szaruhártya, szemlencse és retinaeltéréseket okozhat. A szem eltérései közül gyakori a visszatérő corneaerózió, az anterior lenticonus, a perifériás retinán megjelenő pigmentzavar: a „dot and fleck” retinopathia.Esetismertetés: 38 éves férfi betegünk két hónapja tartó foltlátás miatt jelentkezett klinikánkon. Anamnézisében krónikus glomerulonephritis talaján kialakult veseelégtelenség miatt vesetranszplantáció és hipertónia szerepelt. Szemészeti vizsgálata során békés elülső szegmentum mellett a lencsében lenticonust nem láttunk, a retina középperifériáján „dot and fleck” retinopathiát nem találtunk, de a makulában pigmentegyenetlenséget, pigmentzavart, atrófiát észleltünk. OCT-felvétel a makula atrófiáját megerősítette. FLAG-vizsgálat a makula területében hyperfluoreszcens pontszerű festékhalmozódást mutatott, szivárgás nélkül. A beteg szemészeti gondozásba vételét követően, mindkét oldali fülzúgás és hallásromlás miatt fül-orr-gégészeti vizsgálatra jelentkezett, amely során kétoldali szenzorineurális halláscsökkenést állapítottak meg. Ezzel teljessé váltak az Alport-szindróma tünetei.
Következtetés: A gyakori corneaerózió, anterior lenticonus, és a „dot and fleck” retinopathia mellett, a makuláris atrófia az Alport-szindróma szemészeti eltéréseinek egy ritkábban előforduló formája, amelynek ismerete fontos a látóélesség prognózisának megítélése céljából.
Keywords
Alport syndrome, lenticonus, macular atrophy
Kulcsszavak
Alport-szindróma, lenticonus, makulaatrófia
Bevezetés
Az Alport-szindróma a bazálmembrán IV. típusú kollagénjének örökletes zavara. A IV. típusú kollagén a hálózatképző kollagének csoportjába tartozik, amely biztosítja a bazálmembrán stabilitását, integritását. Amennyiben a COL4A5 (X-hez kötött öröklés esetén) vagy a COL4A3 és a COL4A4 (autoszomális recesszív öröklés esetén) génekben mutáció jön létre, akkor a testszerte megtalálható bazálmembrán-funkció romlása alakul ki (1, 3). Az érintett IV. típusú kollagén megtalálható a vese glomerulusaiban, a cochleában, a cornea Bowman-membránjában, a szemlencse tokjában, a retinában: az idegrost-rétegben, a membrana limitans internában, a pigmentepitheliumban, a Bruch-membránban. Előfordulásának megfelelően a IV. típusú kollagén zavara progresszív vesekárosodást, szenzorineurális halláscsökkenést, és sokrétű ocularis eltéréseket okoz (1, 2). Az Alport-szindróma incidenciája 1/5000 élve született csecsemőre viszonyítva (3).
Az alábbiakban az Alport-szindróma gyakoribb szemészeti megjelenési formáit foglaljuk össze.
Cornea
A visszatérő corneaeróziók, amelyek az X-hez kötött Alport-szindróma esetén kb. 10%-os gyakorisággal fordulnak elő, a Bowman-membrán diszfunkciójára vezethetőek vissza: megfelelő struktúrájú kollagén hiányában a membrán gyenge, nem tapad kellő mértékben az epitheliumhoz. Jellemzően olyan betegekben fordul elő, akiknél korai vesekárosodás figyelhető meg (1).
Lencse
A lencsében létrejövő anterior lenticonust (1. ábra), a lencse középső részének kúp alakú elődomborodását, az elülső lencse tokjának gyengesége okozza; megjelenése Alport-szindrómára diagnosztikus értékű (9). Az X-hez kötött szindrómákban 50% gyakorisággal fordul elő. Az elülső lencsetok sebezhetősége miatt spontán rupturák alakulhatnak ki, amelyek kataraktaképződéshez vezetnek (1). A törési myopia és a szövődményes lencsehomály miatt kialakult látóélesség-romlás intraocularis műlencse-beültetésével javítható (1, 4, 5, 10).
A műtét azonban a lencsetok törékenysége, és megnövekedett rugalmassága miatt kiemelt óvatosságot igényel. Femtolézer alkalmazásával azonban jó eredményeket értek el: a módszer lehetővé teszi a capsulorhexis precízebb kivitelezését, így kikerülhető a tok sérülékenyebb része, és az esetleges spontán tokruptura (4, 5, 10).
Retina
A membrana limitans interna rendellenessége „dot and fleck” (retina pettyezettsége) retinopathiát okozhat a periférián (70% gyakoriság X-hez kötött formában), illetve a középperiférián (80% gyakoriság X-hez kötött formában), megjelenésük igen szenzitív Alport-szindrómára. Az elváltozások a látóélességet nem befolyásolják, terápiát nem igényelnek (1, 9). A corneaeróziókhoz hasonlóan, azonban a retina eltérései is gyakrabban fordulnak elő korai vesefunkció-romlással együtt (1).
A Bruch-membrán és a membrana limitans interna anomáliája együtt járul hozzá a temporális retina elvékonyodásához (55% X-hez kötött formákban). A manifesztáció szenzitivitása Alport-szindrómára kisebb, mind a perifériás „dot and fleck” retinopathiának, a látóélességet azonban szintén nem befolyásolja (1, 9).
Ezen gyakori eltérésekkel szemben, a szindróma ritkább megjelenési formái okozhatnak tartós progresszív látásromlást (1, 9). Óriás makulalyuk, illetve makuláris atrófia szórványosan került eddig leírásra (3, 6, 7, 11). Ismeretük azonban fontos a látás prognózisának előrejelzéséhez.
Esetismertetés
38 éves férfi betegünk két hónapja tartó foltlátás miatt jelentkezett klinikánkon. Kórtörténetében 10 évvel ezelőtt krónikus glomerulonephritis talaján kialakult veseelégtelenség miatt vesetranszplantáció szerepelt, továbbá hipertónia miatt állt belgyógyászati kezelés alatt. Gyermekkorában kezdődött a vesebetegsége, hematuriával, proteinuriával, majd magas vérnyomás kialakulásával. Genetikai vizsgálata igazolta az Alport-szindróma diagnózisát.
A vesetranszplantáció után szisztémás szteroidkezelést kapott, ekkor is látásromlást tapasztalt, de a gyógyszer elhagyása után panaszai megszűntek.
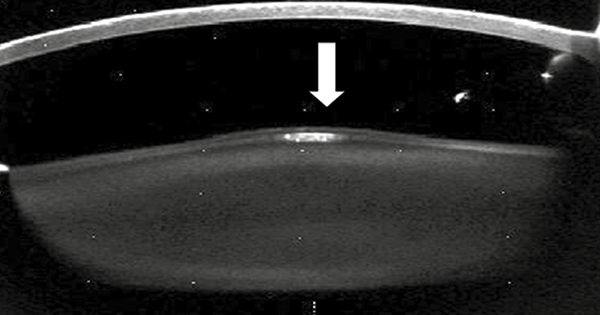
Alport-szindrómában (demonstrációs kép)
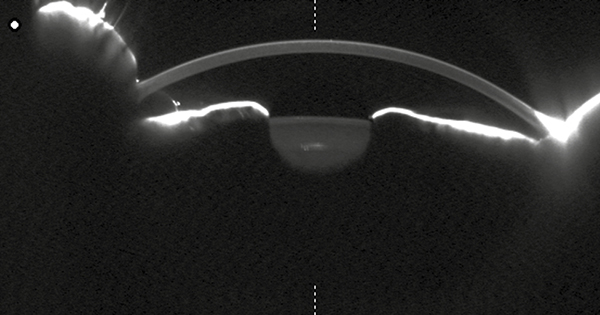
Látóélessége jobb oldalon 1,0, bal oldalon 0,5 volt, közeli vízusa jobb oldalon Cs IV, bal oldalon Cs V + 1,0 Dsph = Cs IV volt, szemnyomás-értékei normáltartományban mozogtak. Szemészeti vizsgálata során a békés elülső szegmentum mellett, a lencsében lenticonust nem találtunk (Scheimpflug képalkotás nem igazolta) (2. ábra), a retina középperifériáján pigmentzavart, „dot and fleck” retinopathiát nem találtunk, azonban a makula területében körkörös pigmentkikopást és pigmentrögöket észleltünk (3. ábra).
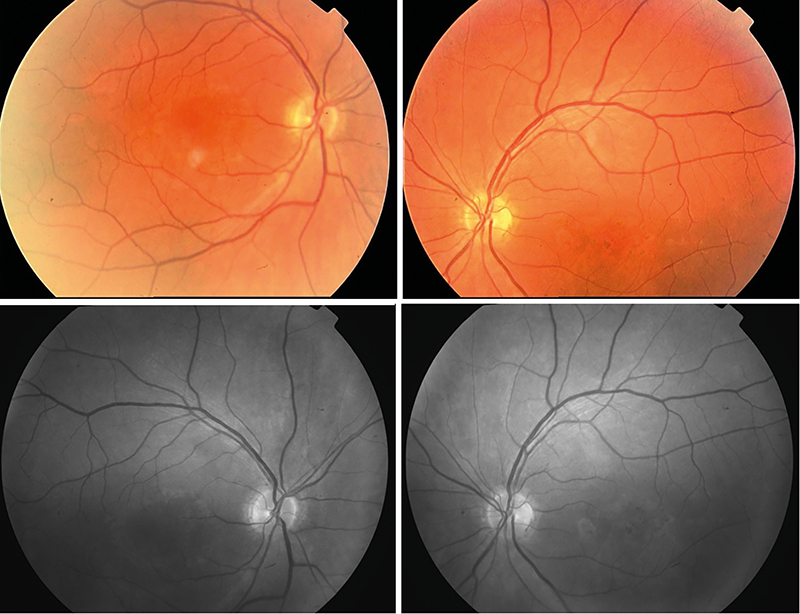
A retina középperifériáján pigmentzavar, „dot and fleck” rethonopathia nem látható, azonban a makula területében körkörös pigmentkikopás és pigmentrögök észlelhetőek szinte szimmetrikusan mindkét oldalon
A makula-OCT-felvételen mindkét oldalon a foveoláris behúzottság elsimult, a retina elvékonyodott (4. ábra).
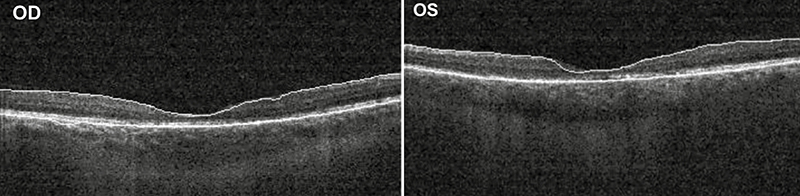
A fovea vastagsága a jobb oldalon 145 mikron, bal oldalon 139 mikron volt. Tekintettel az anamnézisben szereplő vesetranszplantációra, a gondozó nefrológussal történő egyeztetés után fluoreszcein-angiográfiás vizsgálatot végeztünk, amely mindkét oldalon hyperfluoreszcens pontszerű festékhalmozódást mutatott a makula területében, szivárgás nem volt látható (5. ábra).
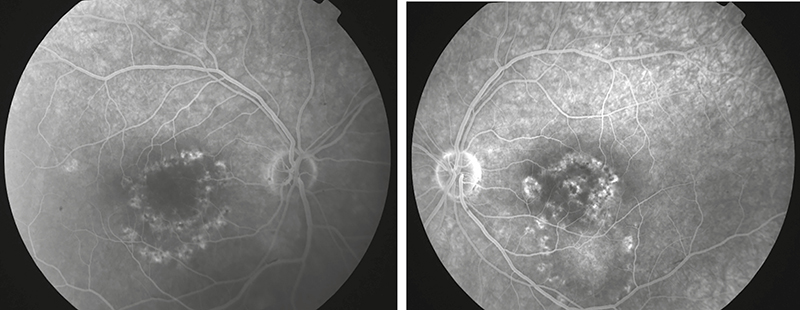
Mindkét funduson a makula területében hyperfluoreszcens, pontszerű festékhalmozódások láthatóak festékszivárgás nélkül. A középperiféria és periféria ép
Elektrofiziológiai vizsgálat csapdisztrófia képét mutatta, ám ez az eltérés kialakulhatott szekunder módon a vesetranszplantációt megelőző hemodialízis következményeként is. Tartós dialízisre szoruló betegeknél Alport-szindróma nélkül is hasonló elektrofiziológiai eltéréseket írtak le (3, 8). A beteget kétoldali tinnitus és halláscsökkenése miatt neurológiai és fül-orr gégészeti kivizsgálásra irányítottuk, ahol audiometria elvégzése után kétoldali szenzorineurális halláscsökkenést állapítottak meg. Így az Alport-szindrómához társuló halláscsökkenése is igazolódott.
A beteget nyomon követtük és fokozatos látásromlását tapasztaltuk. Hat év elteltével legjobb korrigált látóélessége jobb oldalon 0,8, bal oldalon pedig 0,32 volt. A kontroll makula OCT-n a fovea további elvékonyodása látszott, jobb oldalon 128 mikron bal oldalon 121 mikron vastagságú foveával.
Megbeszélés
Az Alport-szindróma a kollagén öröklött rendellenessége miatt alakul ki, amely főként a vesét, a szemet és a cochleát érinti. A betegség genetikailag heterogén, az esetek körülbelül 65%-a X-hez kötött, 15%-ban autoszomális recesszív, és 20%-a autoszomális domináns öröklődést mutat. Előfordulhatnak sporadikus esetek is (6).
Az Alport-szindróma által leginkább érintett szerv a vese. A hematuria a legkorábbi klinikai tünet, ami általában gyermekkorban jelenik meg, mint esetünkben is. Később proteinuria és magas vérnyomás alakul ki, majd korai felnőttkorra létrejön a veseelégtelenség. Leányoknál enyhébbek a tünetek és később alakul ki veseelégtelenség. A halláskárosodás a vesefunkciókkal párhuzamosan változik, rosszabbodik és a cochlearis bazálmembrán rendellenessége okozza.
Ocularis eltérések Alport-szindrómában 30-70% között fordulnak elő (6). Gyermekkorban a szemészeti eltérések ritkák, de a kor előrehaladtával gyakoriságuk és súlyosságuk is növekszik. A szemlencse rendellenessége, az elülső vagy ritkábban a hátsó lenticonus patognomikus értékű. Gyakori szemészeti manifesztációk közé tartoznak még a visszatérő corneaeróziók, és a perifériás „dot and fleck” retinopathia, amelyek nem okoznak vissza nem fordítható látásromlást (1, 9). Ritkán corneadisztrófia is kialakulhat.
Az ismertetett esetünk felhívja a figyelmet az Alport-szindróma ritka, de a látást hosszú távon veszélyeztető megjelenési formájára, a makuláris atrófiára. Ennek ismerete a látás prognózisának felállításában jelentős szereppel bír, illetve anterior lenticonus – katarakta létrejötte esetén segít megbecsülni a műlencse-beültetés utáni várható látóélességet. A látás prognózisának előrejelzése kiemelt jelentőségű Alport-szindrómában, hiszen a betegeknek halláskárosodása is van (5).
Az Alport-szindróma szemészeti manifesztációinak felismerése hozzájárulhat a vesebetegség progressziójának megbecsüléséhez, illetve a diagnózishoz felállításához is. A visszatérő corneaeróziók és a „dot and fleck” retinopathia gyakrabban fordulnak elő korai veseelégtelenségben, így jelenlétükben előrejelezhető a vesebetegség progressziójának várható üteme (1, 9). Az anterior lenticonus és a „dot and fleck” retinopathia nagy szenzitivitással rendelkezik Alport-szindrómában, emiatt fennállásuk gyakorlati segítséget nyújthat a diagnózis felállításában különösen abban az esetben, ha a genetikai vizsgálat nem elérhető (1, 9).
Esetünk érdekességét az adja, hogy az Alport-szindróma egy ritkán megjelenő retinalis elváltozásával találkoztunk: a makulában kialakuló atrófiával, a retina centrális elvékonyodásával, amelynek ismerete, diagnosztikája a betegek látóélességének prognózisa szempontjából döntő jelentőségű.
Következtetés
Az Alport-szindróma egy ritka, örökletes, genetikai megbetegedés, amely a veseműködést, a hallást és a látást is károsítja. A típusos szemészeti tünetek, mint az elülső lenticonus és a „dot and fleck” retinopathia segít a társszakmáknak a korai diagnózis felállításában. Vannak azonban ritkán megjelenő okuláris eltérések is például makuláris atrófia, makulalyuk kialakulása, amelyeknél a vesebetegség, illetve hallászavarok irányítják a figyelmet az Alport-szindrómára.
Nyilatkozat
A szerzők kijelentik, hogy az esetismertetésük megírásával kapcsolatban nem áll fenn velük szemben pénzügyi vagy egyéb lényeges összeütközés, összeférhetetlenségi ok, amely befolyásolhatja a közleményben bemutatott eredményeket, az abból levont következtetéseket vagy azok értelmezését.
Irodalom
1. Savinge J, Sheth S, Leys A, et al. Ocular features in Alport syndrome: pathogenesis and clinical significance. Clin J Am Soc Nephrol. 2015; 4: 703–709.
https://doi.org/10.2215/CJN.10581014
2. Savinge J, Liu J, DeBuc DC, et al. Retinal basement membrane abnormalities and the retinopahy of Alport syndrome. Invest Ophthalmol Vis Sci 2010; 3: 1621–1627.
https://doi.org/10.1167/iovs.08-3323
3. Igami TZ, Lavezzo MM, Ferraz DA, et al. Unusual macular thickness in Alport syndrome: case report. Arq Bras Oftalmol 2012; 75: 283–285.
https://doi.org/10.1590/s0004-27492012000400014
4. Ecsedy M, Sándor GL, Takács ÁI, et al. Femtosecond laser-assisted cataract surgery in alport syndrome with anterior lenticonus. Eur J Ophthalmol 2015; 25: 507–511.
https://doi.org/10.5301/ejo.5000603
5. Ecsedy M, Nagy ZZ. Reply to: Cataract surgery in patienst with Alport syndrome. Eur J Ophthalmol 2016; 26: e86.
https://doi.org/10.5301/ejo.5000775
6. Sargazi M, Dehghani S, Dahmardeh M, et al. Ocular Manifestations of Alport Syndrome: Report and Comparison of Two Cases. Cureus 2023; 15: e47373.
https://doi.org/10.7759/cureus.47373
7. Panchal B, Doshi S, Pathengay A. Unilateral giant macular hole in a case of Alport syndrome. Indian J Ophthalmol 2019; 67: 1731.
https://doi.org/10.4103/ijo.IJO_650_19
8. Jeffrey BG, Jacobs M, Sa G, et al. An electrophysiological study on children and young adults with Alport's syndrome. British Journal of Ophthalmology 1994; 78: 44–48.
https://doi.org/10.1136/bjo.78.1.44
9. Wang D, Pan M, Li H, et al. Four novel mutations identified in the COL4A3, COL4A4 and COL4A5 genes in 10 families with Alport syndrome. BMC Med Genomics 2024; 17: 181.
https://doi.org/10.1186/s12920-024-01953-0
10. Gouws D, van der Westhuizen DP, Stuart KV, et al. Bilateral anterior lens capsule rupture in Alport syndrome: case series and literature review Digit J Ophthalmol 2024; 30: 55–59.
https://doi.org/10.5693/djo.02.2024.03.001
11. Dolz-Marco R, Gallego-Pinazoa R, Francés-Munoza B, et al. New macular tomography findings in Alport syndrome. Arch Soc Esp Oftalmol 2012; 87: 55–56.
https://doi.org/10.1016/j.oftal.2011.09.004