A case of a child diagnosed with X-linked lymphoproliferative disease in whom Epstein–Barr virus infection led to bilateral retinal necrosis
doi: 10.55342/szemhungarica.2026.163.2.70
Case report
Summary
X-linked lymphoproliferative XLP) is a rare condition associated Epstein–Barr virus (EBV) infection is widespread in the adult population, typically causing mild symptoms in children and presenting as mononucleosis in adolescents and adults. In XLP, EBV infection triggers a fulminant immune response. The clinical manifestations and course of the disease can vary significantly. There is very limited literature available regarding ophthalmic complications associated with XLP. The purpose of this case report is to highlight potential ophthalmic consequences and present treatment options. The authors describe the case of a 5-year-old boy in whom genetic testing confirmed haemophagocytic lymphohistiocytosis, which in this case, led to secondary glaucoma and bilateral retinal necrosis. The patient's ophthalmic symptoms showed regression following local and systemic steroid and antiviral therapy.
Összefoglaló
Az X-kromoszómához kötött lymphoproliferativ betegség (XLP) egy ritka, immundeficienciával járó kórkép. Az Epstein–Barr-vírus (EBV) fertőzés igen elterjedt a felnőtt populációban, gyermekek esetében rendszerint enyhe tüneteket okoz, serdülők és felnőttek esetében mononucleosis képében jelentkezik. XLP esetén az EBV-fertőzés fulmináns immunreakciót indít el. A betegség által okozott klinikai tünetek és azok lefolyása igen változatos lehet. Kevés irodalmi adat áll rendelkezésünkre az XLP-vel kapcsolatos szemészeti szövődményekről. Esetismertetésünk célja, hogy felhívjuk a figyelmet a potenciális szemészeti következményekre és a kezelési lehetőségekre. Egy 5 éves fiúgyermek esetét ismertetjük, akinél genetikai vizsgálattal igazolt XLP talaján EBV indukált hemofagicitás limfohisztiocitózis zajlott, amely a látószerveben szekunder glaukómát, valamint kétoldali retinanekrózist okozott. A beteg szemészeti tünetei lokális valamint szisztémás szteroid- és antivirális terápia mellett regressziót mutattak.Keywords
Epstein–Barr virus, X-linked lymphoproliferative disease, immunodeficiency, retinal necrosis, secondary glaucoma
Kulcsszavak
Epstein–Barr-vírus, X-kromoszómához kötött lymphoproliferativ betegség, immundeficiencia, retinanekrózis, szekunder glaukóma
Bevezetés
Az X-kromoszómához kötött lymphoproliferativ betegség (XLP) egy ritka, súlyos immundeficienciával járó genetikai kórkép, amely leggyakrabban az Epstein–Barr-vírus (EBV) fertőzés kapcsán válik klinikailag nyilvánvalóvá (1). Az EBV a herpeszvírusok családjába tartozó gamma-herpesz-vírus (2). A felnőtt populáció 90-95%-a átfertőződést mutat a vírussal (3). Az EBV a szem minden szegmensében okozhat manifesztációt. Okozhat dacryoadenitist, az elülső szegmes érintettsége lehet conjunctivitis, episcleritis, keratitis és iritis. Hátsó szegmens érintettsége lehet uveitis, retinitis, opticus neuritis (4). Az XLP esetében a beteg immunrendszere nem képes megfelelően reagálni az EBV-fertőzésre, ami fulmináns immunreakciókhoz, hemofagocitás lymphohistiocytosishoz (HLH), lymphomához, valamint egyéb szervi szövődményekhez vezethet (1). Incidenciája 1-3 az 1 millióból (5). Az XLP X-kromoszómához kötött módon öröklődik. Kialakulásának oka lehet az SH2D1A génmutációja, amely a slam associated proteint (SAP) kódolja, illetve az X-kapcsolt apoptózis inhibitor fehérje (XIAP) mutáció. Azok a fiúgyermekek, akik öröklik a patogén variánst érintettek lesznek, a leány gyermekek, akik öröklik a patogén variánst, heterozigóták lesznek, és általában nem érintettek, ritka esetben X-kromoszóma-inaktiváció esetében a heterozigóta nők is mutathatnak tüneteket (1). Klinikai megjelenését tekintve elkülönítjük két fenotípusát: a hemofagocitás lymphohistiocytosist és a lymphoproliferációt.
A betegség szemészeti manifesztációiról kevés irodalmi adat áll rendelkezésre. Az EBV-fertőzéssel összefüggésben kialakuló szövődmények, például a bilaterális retinanekrózis, ritka, de súlyos következmények lehetnek. Ezek a szemészeti tünetek a látás jelentős romlásához vagy akár teljes elvesztéséhez is vezethetnek, amennyiben nem ismerik fel és kezelik időben.
A következő esetismertetés célja, hogy felhívja a figyelmet az XLP szemészeti szövődményeire, bemutatva egy 5 éves fiúgyermek esetét, akinél a betegség kétoldali retinanekrózist és szekunder glaukómát okozott. Az időben alkalmazott szisztémás antivirális és gyulladáscsökkentő terápia jelentősen javította a beteg klinikai állapotát, és rámutatott a korai diagnózis és kezelés kiemelt fontosságára az XLP-vel összefüggő szemészeti szövődmények esetében.
Szisztémás tünetek és kezelésük
Az első megjelenéskor 4 éves fiúgyermek kórházi ellátására 2 hete tartó lázas állapot, második héttől testszerte jelentkező exanthemák, lymphadenopathia miatt került sor. Felvételekor mononucleosis szindrómára jellemző klinikai kép volt látható. Laborleleteiben emelkedett nekroenzimszint mutatkozott, amely ismételt laborvizsgálata során tovább emelkedett, valamint thrombocytopenia, hypoalbuminaemia és hyponatraemia jelentkezett. Hasi ultrahangvizsgálata hepatosplenomegaliát, ödémás falú epehólyagot írt le. Ápolásának 7. napján több alkalommal görcstevékenysége zajlott, amely diazepam, majd midazolam adását követően oldódott. Koponya MR-vizsgálata érdemi eltérést nem igazolt. Zavart tudati állapot, valamint hányások miatt intubálásra került. Magas oxigénigénye miatt mellkas-röntgenvizsgálat készült, amelyen kétoldali pneumónia ábrázolódott, amely miatt ceftriaxonterápia indult. Ferritin meghatározása mérhetetlenül magas szintet mutatott. Romló általános állapota hátterében hemofagocitás lymphohistiocytosis merült fel. Virológiai vizsgálata a feltételezett akut EBV-infekciót megerősítette (EBV IgM-pozitív, 21800 U/ml). Dexametazon- és IVIG-terápia indult, valamint fibrinogén, thrombocyta és 1E koncentrált vvt-pótlásban részesült. A betegség második hetében az infektológiai osztályról intenzív osztályra helyezték át. Immunszupprimált állapota miatt trimethoprim-sulfomethoxazol és fluconazol-profilaxisban részesült. Keringéstámogatásként noradrenalint igényelt. Klinikai tünetei kielégítették a HLH diagnosztikus kritériumait. A HLH 2004. évi terápiás protokollja szerint dexametazonnal, etoposiddal, intrathecalis methotrexáttal, intrathecalis prednisolonnal, cyclosporinnal, anakinrával, intravénás és intrathecalis rituximabbal folytatódott kezelése. Csontvelői mintavétel és lumbálpunkció is történt. A liquorban magas kópiaszámmal EBV jelenléte igazolódott, HLH idegrendszeri manifesztációja mellett. A csontvelői mintában hypocellularis csontvelő, hemofagocitózis volt látható. A szövettani vizsgálat és a liquorvizsgálat igazolta az idegrendszeri érintettséggel járó hemofagocitás lymphohistiocytosist. Az EBV-fertőzés igazolása után ganciclovir kezelése indult. Oligo-anuriája kezdetben diuretikummal, később folyamatos vesepótló kezeléssel került ellátásra. Polyserosistis alakult ki perikardiális, pleurális és szabad hasi folyadékkal, ami lélegeztetését tovább nehezítette. Keringéstámogatás segítésére noradrenalin mellett 6 napig inodilatátor (milrinon) került alkalmazásra. Kiugró lipáz és amiláz laborértékei hátterében pancreatitis igazolódott, amely konzervatív terápia mellett javult. Invazív lélegeztetésre összesen 10 napig, noninvazív légzéstámogatásra 4 napig volt szüksége. A kezelés mellett gyulladásos paraméterei és az EBV kópiaszám csökkenő tendenciát mutatott. Genetikai vizsgálata kóroki SH2D1A mutációt igazolt. A súlyos klinikai tünetek miatt allogén csontvelő-transzplantációra került sor.
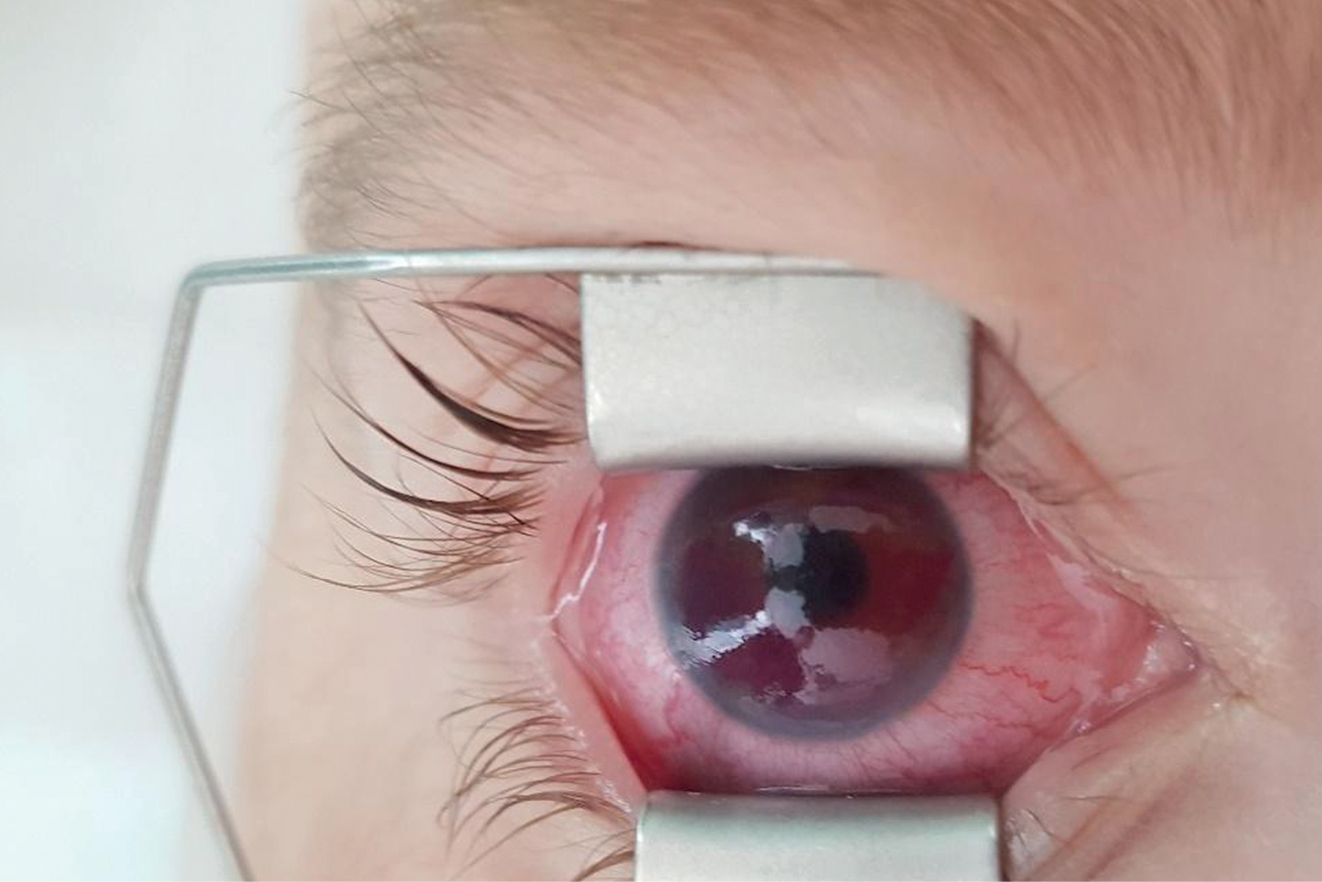
Szemészeti tünetek és kezelésük
Röviddel a csontvelő-transzplantációt követően észlelték a jobb szem kötőhártya belövelltségét, emiatt kérték első szemészeti vizsgálat elvégzését az intenzív osztályon.
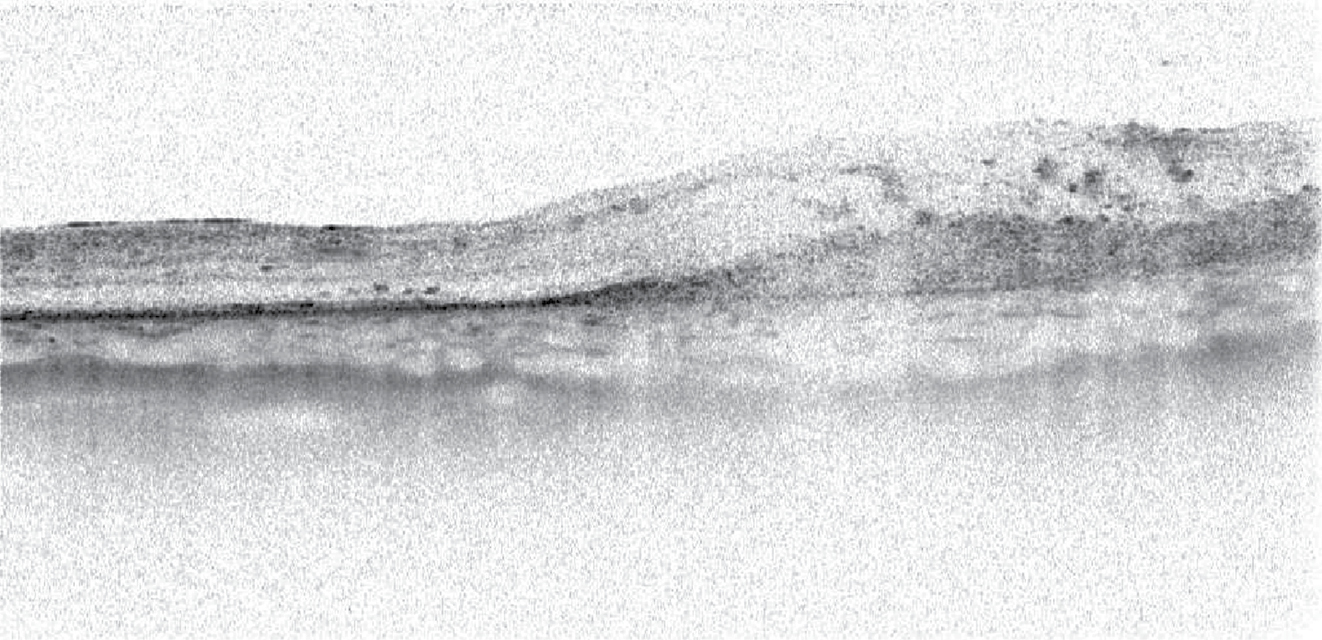
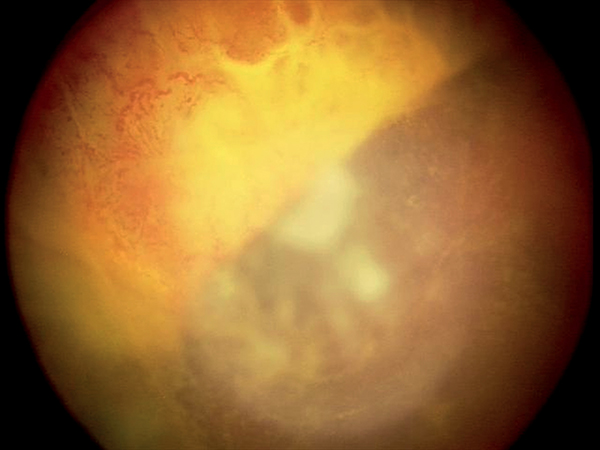
Szemészeti státuszában a jobb szemen iris-neovaszkularizáció, tág, fénymerev pupilla látszott, vörös visszfény nem volt nyerhető, érdemi fundusvizsgálat itt nem volt lehetséges, ultrahangvizsgálattal vérzéses chorioidea-ablatio jelei ábrázolódtak. A szemnyomás 58 Hgmm volt Icare tonométerrel mérve. A bal oldalon a szemfenéki kép retinochorioiditis típusos képét mutatta: sárgásfehér kiterjedt retinális beszűrődéssel, iszkémiás területekkel, makulatáji vérzéssel és ödémával.
Lokális terápiaként mindkét szembe naponta 4× ganciclovir 1,5 mg/g szemgélt (Virgan), 3×1 dexametazon 1 mg/ml (Maxidex) cseppet és diclofenac 1 mg/ml (VoltarenOphtha) szemcseppet kapott. A jobb szem kezelése az extrém magas szemnyomás csökkentése céljából naponta 2×1 dorzolamid/timolol 20 mg/ml + 5 mg/ml (Cosopt Multi) szemcseppel egészült ki. Szisztémásan a fentiek szerint ismertetett antivirális és immunszuppresszív kezelése zajlott. Másfél évvel a betegség lezajlása után a jobb szemen a látóélesség fényérzés nélküli volt, bal szemen 0,12 ETRDS táblán.
Megbeszélés
A HLH-nak két típusát különítjük el: elsődleges és másodlagos formát.
Az elsődleges, autoszomális recesszív forma, amelyet familiáris hemofagocitás lymphohistiocytosisnak neveznek. Előfordulásának becsült aránya körülbelül 1:50 000. Az autoszomális recesszív öröklődésmenetre tekintettel a családi anamnézis gyakran negatív. Az első megjelenése jellemzően csecsemőkorban történik, de serdülők és felnőttek esetében is előfordulhat.
A másodlagos HLH (sHLH) akkor alakul ki, amikor valamilyen erős immunológiai aktiváció történik a szervezettel, például egy súlyos fertőzés válthatja ki. Ezt a formát gyakran lehet megfigyelni immunhiányos állapotú betegeknél, különösen vírusfertőzések kapcsán. Ezen kívül az sHLH előfordulhat daganatos betegségek során is, amikor vagy a betegség első klinikai megjelenése lehet a HLH, vagy a daganatos kezelés során alakul ki (6).
A hemofagocitikus szindróma patomechanizmusát a Th1 proinflammatorikus citokinek diszfunkciója által okozott állapot váltja ki. Jellemzője a Th1 profilhoz tartozó citokinek túltermelődése, beleértve az interferon-gammát, a tumornekrózis-faktor-alfát, az interleukin (IL)-6-ot, IL-10-et, IL-12-t, valamint az sIL-2 R-t (CD25). Ez a T-lymphocyták és makrofágok aktivációját eredményezi (7).
A HLH klinikai tünetei igen szerteágazóak lehetnek, több szervrendszert is érintenek.
A HLH leggyakoribb tünetei a láz, hepatosplenomegalia és cytopeniák. Gyakran megfigyelhető továbbá hypertrygliceridaemia, coagulopathia hypofibrinogenemiával, májfunkciós zavarok, emelkedett ferritin- és szérum transzaminázszintek. Neurológiai érintettség, liquor hyperproteinaemiával és mérsékelt pleocytosissal. Egyéb, ritkább kezdeti klinikai jelek közé tartozik a lymphadenopathia, bőrkiütés, sárgaság és ödéma. Az általunk ismertetett esetben a sárgaságon kívül az összes fenti klinikai tünet megjelent betegünk követése során.
A kórszövettani vizsgálatok során lymphocyták és érett makrofágok felszaporodása figyelhető meg, melyet esetenként hemofagocytosis kísér. Ez elsősorban a lépben, a megnagyobbodott nyirokcsomókban, a csontvelőben, a májban és a cerebrospinális folyadékban figyelhető meg. A HLH egyéb gyakori laboratóriumi eltérései közé tartozik a csökkent természetes ölősejt (NK) aktivitás, valamint a fokozott citokin termelés, utóbbi különösen az oldott interleukin-2 receptor (sIL-2R) emelkedett szérum- és liquor szintjében nyilvánul meg (6).
Luping és munkatársai által 2023-ban publikált retrospektív tanulmány 1525 HLH-beteg adatait dolgozta fel a szemészeti manifesztációt illetően, 341 betegnél történt szemészeti vizsgálat és 133 betegnél találtak szemészeti eltérést. A leggyakoribbak a hátsó szegmens betegségei voltak (66 beteg, 49,62%), ideértve a retina- és üvegtesti vérzést, a serosus retinaleválást, a cytomegalovírus-retinitist és a papillaödémát. Egyéb, megfigyelt szemészeti komplikációk közé tartoztak a szemfelszíni fertőzések (kötőhártya-gyulladás, 34 beteg, 25,56%; keratitis, 16 beteg, 12,03%), a subconjunctivalis vérzés (11 beteg, 8,27%), a chemosis (5 beteg, 3,76%), az elülső uveitis (11 beteg, 8,27%), a szteroidindukált glaukóma (5 beteg, 3,76%), a sugárzás okozta szürkehályog (1 beteg, 0,75%), a dacryoadenitis (2 beteg, 1,50%), a dacryocystitis (1 beteg, 0,75%), az orbitacellulitis (2 beteg, 1,50%), az orbita-pseudotumor (2 beteg, 1,50%) és a kancsalság (2 beteg, 1,50%) (8).
Esetünkben a beteg szemészeti vizsgálatakor retinanekrózis volt látható a bal szemen. Ennek hátterében leggyakrabban herpeszvírus által okozott fertőzés áll, gyakorisági sorrendben HSV1, VZV, HSV2, CMV. Az EBV-fertőzést extrém ritkán, de igazolták az akut retinanekrózis (ARN) hátterében (9). A szemérintettség gyakoribb központi idegrendszeri érintettség esetén, amely betegünk esetében is zajlott (10). A liquor PCR-vizsgálata igazolta az EBV központi idegrendszeri jelenlétét. Diagnosztikailag nehézséget jelentett, hogy a jobb szemen chorioidea-ablatio állt fenn, így üvegtesti mintavétel csak az egyetlen látó szemből lett volna lehetséges. A mintavétel mellett szólt az EBV-fertőzés intraocularis jelenlétének igazolása, amely esetben adható lett volna ganciclovirkezelés. Az üvegtesti mintavétel kockázata betegünk esetében igen magas volt, tekintettel a beteg egyetlen látásmaradvánnyal bíró szemére, alacsony életkorára, az atrófiás retinaterületek miatti fokozott retinaleválás rizikóra, thrombocytopenia miatti fokozott vérzésrizikóra, és az immunhiányos állapot jelentette fokozott fertőzéshajlamra. A csarnokvízből történő mintavétel esetén a vizsgálat szenzitivitása ismerten alacsonyabb. A már bizonyítottan zajló szisztémás és KIR-i EBV-infekció esetén erősen alátámasztható volt az EBV kóroki szerepe a szemészeti tünetek mögött is. Intenzív osztályos kezelőorvosával, illetve a szülőkkel történt egyeztetés után esetünkben a fenti mérlegelést követően nem végeztünk intraocularis mintavételt. A fent ismertetett terápia mellett javulást tapasztaltunk, amely „ex iuvantibus” a kóroki gyanút támasztotta alá.
Következtetések
A szerzők egy ritka, X-kromoszómához kötött lymphoproliferativ betegségben szenvedő 5 éves fiúgyermek esetét ismertették, akinél az Epstein–Barr-vírus fertőzés következtében kétoldali retinanekrózis és szekunder glaukóma alakult ki. Az XLP egy súlyos immunhiányos állapot, amely az EBV-re adott kóros immunválasz miatt fulmináns tünetekkel és szervi károsodásokkal járhat. Betegünk intenzív osztályos kezelést és célzott terápiát igényelt. Szemészeti szempontból a kétoldali retinanekrózis és az ehhez társuló szövődmények súlyos látáskárosodás kockázatát hordozták. A helyi és szisztémás antivirális, valamint gyulladáscsökkentő kezelés hatására a beteg szemészeti állapota javulást mutatott. Az eset hangsúlyozza az XLP-vel kapcsolatos szemészeti szövődmények felismerésének és időbeni kezelésének jelentőségét, valamint rávilágít a multidiszciplináris megközelítés szükségességére az ilyen komplex kórképek ellátásában.
Anyagi támogatás
A témához kapcsolódó poszter az European Paediatric Ophthalmological Society 2023. évi kongresszusán került bemutatásra, a részvételt a Magyar Gyermekszemészek és Strabológusok Társasága támogatta.
A közlemény más folyóiratban korábban nem jelent meg, máshova beküldésre nem került.
Rövidítések:
EBV: Epstein–Barr-vírus; ETRDS: Early Treatment Diabetic Retinopathy Study; HLH: hemofagocitás limfohisztiocitózis; IVIG: intravénás immunglobulin; SAP: slam associated protein; sHLH: szekunder hemofagocitás limfohisztiocitózis; vvt: vörösvértest; XIAP: X-linked inhibitor of apoptosis protein/X-kapcsolt apoptózis inhibitor fehérje; XLP: X-kromoszómához kötött lymphoproliferativ betegség/X-linked lymphoproliferative disease
Irodalom
1. Meyer L, Hines M, Zhang K, et al. X-Linked Lymphoproliferative Disease, in GeneReviews(®), Adam MP, Feldman J, Mirzaa GM, et al. Editors. 1993, University of Washington, SeattleCopyright ©
https://doi.org/10.1007/springerreference_40035
2. Odumade OA, Hogquist KA, and Balfour HH, Jr. Progress and problems in understanding and managing primary Epstein-Barr virus infections. Clin Microbiol Rev 2011; 24: 193–209.
https://doi.org/10.1128/cmr.00044-10
3. Wong Y, Meehan MT, Burrows SR, et al. Estimating the global burden of Epstein–Barr virus-related cancers. Journal of Cancer Research and Clinical Oncology 2022; 148: 31–46.
https://doi.org/10.1007/s00432-021-03824-y
4. Matoba AY Ocular disease associated with Epstein-Barr virus infection. Surv Ophthalmol 1990; 35: 145–50.
https://doi.org/10.1016/0039-6257(90)90069-8
5. Meazza R, Tuberosa C, Cetica V, et al. Diagnosing XLP1 in patients with hemophagocytic limfohisztiocitózis. Journal of Allergy and Clinical Immunology 2014; 134: 1381–1387.e7.
https://doi.org/10.1016/j.jaci.2014.04.043
6. Henter JI, Horne A, Aricó M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007; 48: 124–31.
https://doi.org/10.1002/pbc.21039
7. Suhr KS, Chiang MF, Flynn JT, et al. Ocular involvement in hemophagocytic syndrome: a novel funduscopic manifestation and review of the literature. Retin Cases Brief Rep 2016; 10: 345–8.
https://doi.org/10.1097/icb.0000000000000255
8. Wang L, Suo L, Kou F, et al. Ocular Phenotypes in Patients With Hemophagocytic Lymphohistiocytosis: A Retrospective Analysis in a Single Center Over 7 Years. Am J Ophthalmol 2023; 253: 119–131.
https://doi.org/10.1016/j.ajo.2023.05.011
9. Soitong P, Ngathaweesuk Y, Panyayingyong N, et al. Multicenter Analysis of Acute Retinal Necrosis: Clinical Characteristics, Viral Pathogens, and Diagnostic Predictive Factors. Ocul Immunol Inflamm 2025; 1–8.
https://doi.org/10.1080/09273948.2025.2456642
10. Dutz JP, Benoit L, Wang X, et al. Lymphocytic vasculitis in X-linked lymphoproliferative disease. Blood 2001; 97: 95–100.
https://doi.org/10.1182/blood.v97.1.95