Genotype-phenotype correlations in USH2A gene mutation-related RP presented with our genotyped cases
doi: 10.55342/szemhungarica.2022.159.3.130
Original article
Summary
Introduction: Usher syndrome is an autosomal recessive hereditary disease that ophthalmologically manifests itself in symptomatic retinitis pigmentosa. Usher syndrome has three major types. Along with sight loss, hearing loss is a typical symptom that appears during infancy in type I of the disease and during school age in type II. In type III, besides sight and hearing loss, balance disorder is also characteristic.
In type II, the significant narrowing of the visual field develops in the third decade, followed by the damage of colour vision and central sight. In the background of its pathogenesis, biallelic mutations of ADGRV1, USH2A and WHRN genes have been reported. Our aim is to precisely assess patients with hearing loss and retinitis pigmentosa, as well as to perform their genotyping.
Materials and methods: 2 male and 2 female patients were examined at the Department of Ophthalmology of Semmelweis University with standard methods and multimodal imaging. Genotyping of patients with characteristic symptoms was performed with new generation sequencing (NGS, Illumina NextSeq, Usher Targeted Panel). The validation of the variants was done with Sanger sequencing.
Results: We identified four patients with type II Usher syndrome who all had the biallelic mutation of USH2A gene.
Conclusion: With detailed clinical, imaging and electrophysiological examinations as well as with genotyping, we documented both the phenotype and the genotype of our patients. We have observed that the severe phenotype is caused by mutations involving changes in DNA sequence length.
Összefoglaló
Bevezetés: Az Usher-kór autoszomális recesszív módon öröklődő betegség, amely szemészeti szempontból szindrómás retinitis pigmentosa képében manifesztálódik. Az Usher-szindrómának három fő típusa ismert. A látásromlás mellett a halláscsökkenés a jellemző tünet, amely az I. típusban, csecsemőkorban, a II. típusban kisiskoláskor körül jelenik meg. A III. típusában a hallás- és látásromlás mellett egyensúlyprobléma fordul elő.
A II. típusban a látótér jelentős beszűkülése általában a húszas években alakul ki, amelyet a színlátás és a centrális látás megromlása követ. A betegség kialakulásának hátterében az ADGRV1, USH2A, és a WHRN gének biallélikus mutációi ismertek. Célunk a hallásromlással és retinitis pigmentosára jellemző tünetekkel jelentkező betegek részletes szemészeti funkcionális és eszközös vizsgálata, valamint genotipizálása volt.
Anyag és módszer: A Semmelweis Egyetem Szemészeti Klinikáján működő szemészeti genetikai szakrendelésen jelentkező betegek közül 2 férfit és 2 nőbeteget vizsgáltunk standard szemészeti vizsgálómódszerekkel és multimodális képalkotókkal. A jellemző klinikai képet mutató páciensek genotipizálása új generációs szekvenálással (NGS, Illumina NextSeq készülék, Usher Targeted Gene Panel), a variánsok validálása Sanger-szekvenálással történt.
Eredmények: A vizsgálatban 4 Usher-szindróma II-es típusában szenvedő beteget sikerült identifikálni, minden esetben az USH2A gén biallélikus mutációi igazolódtak.
Következtetés: A részletes klinikai, képalkotó és elektrofiziológiai vizsgálatokkal, illetve a genotipizálás elvégzésével mind a fenotípus, mind a genotípus dokumentálása megtörtént. Megfigyeltük, hogy a súlyos fenotípus hátterében a DNS-szekvencia hosszának megváltozásával járó mutációk állnak.
Keywords
Usher, genotype, phenotype, OCT, FAF
Kulcsszavak
Usher, genotípus, fenotípus, OCT, FAF
Bevezetés
Az Usher-kór autoszomális recesszív módon öröklődő betegség, amely szemészeti szempontból szindrómás retinitis pigmentosa képében manifesztálódik. Az Usher- szindrómának három fő típusa ismert. A látásromlás mellett a halláscsökkenés a jellemző tünet, amely az I. típusban, csecsemőkorban, a II. típusban, iskoláskorban jelenik meg. A III. típusában a hallás- és látásromlás mellett egyensúlyprobléma fordul elő (1-4).
A betegség első típusában több gén (MYO7A, USH1C, USH1G, CDH23, PCDH15, CIB2) mutációja is érintett, amelyek fehérjetermékei többek között sejtadhéziós, „scaffold”, valamint ionkötő szerepet látnak el. A betegek veleszületett halláskárosodása súlyos vagy teljes. A szimptómás retinitis pigmentosa tünetei pubertáskorban kezdődnek, a diagnózisra jellemzően ebben az életkorban kerül sor, a csökkent látás („legal blindness”) pedig általában a negyedik évtizedben alakul ki. A betegséget emellett vesztibuláris hipofunkció és megkésett motoros fejlődés is jellemzi (1-4).
A szindróma második típusában az USH2A, WHRN és az ADGRV1 gének mutációi ismertek, ezen gének sejtadhéziós és „scaffold” proteinek mellett G-fehérje kapcsolt receptort kódolnak. A veleszületett halláskárosodás közepes mértékű vagy súlyos, és jellemzően a magas frekvenciákat érinti normális vesztibuláris funkció mellett. A szemészeti tünetek tizenévesen kezdődnek, a retinitis pigmentosa diagnózisát általában a húszas évek során állítjuk fel; a jogi értelemben vett vakság pedig rendszerint a hatodik évtizedben fordul elő (1-4).
A hármas típus jelentősen ritkább, az eltéréseket a jelenleg ismeretlen funkciójú CLRN1 gén mutációja okozza, az általa kódolt clarin 1-fehérje egy transzmembrán-protein. A halláscsökkenés a beszédtanulás után alakul ki; a hallást, a látást és a vesztibuláris rendszert érintő tünetek súlyossága igen nagy változatosságot mutat. A hallásvesztés progresszív jellegű; a vesztibuláris diszfunkció általában enyhe és az esetek 50%-ában fordul elő. A látáspanasz általában nyctalopiával (szürkületi látászavar) kezdődik, jellemző a csőlátótér kialakulása és a második évtizedben jelentkező centrális látásromlás (1–4).
Betegek és módszerek
A vizsgált betegek a Semmelweis Egyetem Szemészeti Klinikáján működő szemészeti genetikai szakrendelésen jelentkező betegek közül 2 férfi és 2 nőbeteg voltak.
Vizsgálati módszereink alapját standard szemészeti vizsgálatok (refrakció, visus, biomikroszkópos vizsgálat fundustükrözéssel, fundusfotó – Topcon© funduskamera) adták. Emellett multimodális képalkotást (IR fundusfotó, OCT, FAF – Spectralis Heidelberg© is alkalmaztunk. Elektrofiziológiai vizsgálataink a „full-field” és a multifokális ERG-t foglalták magukban. A genotípust ismertető genetikai vizsgálatot a Molekuláris es Klinikai Szemészeti Intézet (Basel) laboratóriumában végezték új generációs szekvenálással (NGS, Illumina NextSe© készülék, Usher Targeted Panel), a validálás Sanger-szekvenálással történ.
Eredmények
A vizsgálatban 4 Usher-szindróma II-es típusában szenvedő beteget sikerült azonosítani, minden esetben az USH2A gén biallélikus mutációi igazolódtak.
Első vizsgált betegünk egy 44 éves férfi, aki szindrómás retinitis pigmentosával érkezett. Hallókészüléket ötéves kora óta visel; első szemészeti tünete a 14 éves korában felismert farkasvakság volt. Az Usher-szindróma diagnózisára 26 éves korában került sor. Szemészeti anamnézisében mindkét oldali pseudophakia szerepel, a kataraktaműtétekre 12 év különbséggel került sor. Általános anamnézisében kezelt hipertónia szerepel. Legjobb korrigált visusa vizsgálatakor jobb oldalon fé, bal oldalon kml. Vizsgálata során fotoaverzió jellemezte. Tíz évvel ezelőtti látótérvizsgálata 10°-ra beszűkült látóteret, csőlátást ábrázolt. „Full-field” ERG-vizsgálatán a scotopikus és a fotopikus válasz megszűnt; multifokális ERG-n súlyosan csökkent amplitúdók voltak megfigyelhetők a centrális gyűrűkben. Fundusán réslámpás vizsgálattal porcelánfehér papilla, szűkebb erek, makulatájon céltábla rajzolatú („bull’s eye-like”) megjelenés, a periférián csontsejtszerű és paravasalis pigmentáció volt látható. Emellett fundusfotó és OCT-vizsgálat is készült (1-2. ábrák).
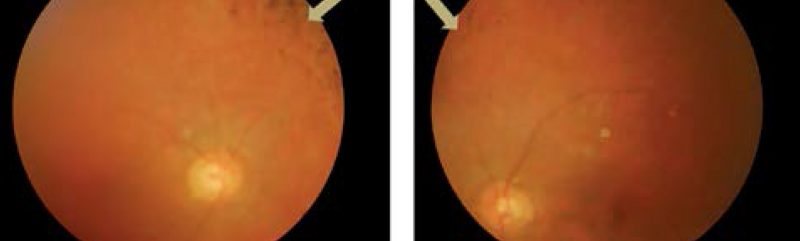
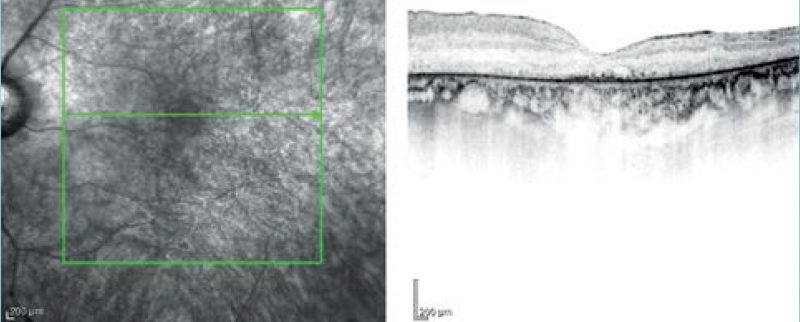
Genetikai eredménye heterozigóta kereteltolódást okozó duplikációt és intronikus mutációt mutatott az USH2A génben (1. táblázat).
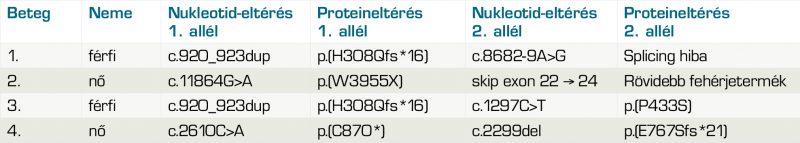
Második betegünk egy 64 éves görög származású nő, szindrómás retinitis pigmentosával. Családi anamnézise pozitív, anyai ágon mind nők, mind férfiak érintettek voltak a betegség tüneteiben. Hallókészüléket gyermekkora óta visel, az Usher-szindróma diagnózisát 39 éves korában kapta meg. Anamnézisében a 90-es években végzett mindkét oldali radiális keratotomia szerepel, valamint 6 évvel ezelőtt mindkét oldali szürkehályog-műtét történt 1 év különbséggel. Általános anamnézisében osteoporosis szerepel. Vizsgálata során fotoaverzió jellemezte. Refrakciója: –4,0 Dsph/–4,5 Dsph; BCVA: 0,1/0,1. Egy évvel ezelőtti látótérvizsgálata csőlátást, 5°-ra beszűkült látóteret ábrázolt. „Full-field” ERG-vizsgálatán a scotopikus és a fotopikus válasz megszűnt; multifokális ERG-n súlyosan csökkent amplitúdók a centrális gyűrűkben. Fundusán réslámpás vizsgálattal halvány papilla, szűkebb erek, atrófiás makulatáj, periférián csontsejtszerű és paravasalis pigmentáció, nagy atrófiás foltok voltak láthatók. Emellett fundusfotó, FAF- és OCT-vizsgálat is készült (3-4. ábrák). Genetikai eredménye heterozigóta korai terminációt okozó „stop mutációt” és deléciót mutációt mutatott az USH2A génben (1. táblázat).

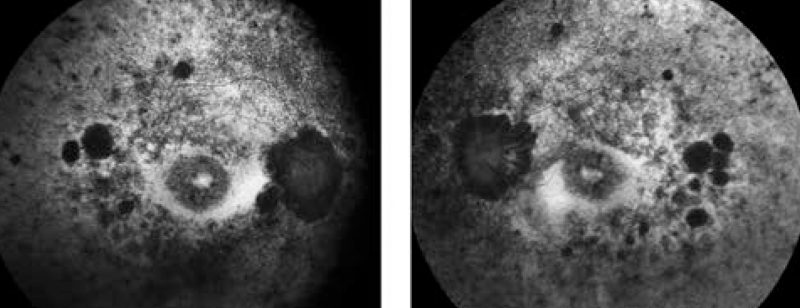
Harmadik betegünk egy 48 éves férfi volt, szintén szindrómás retinitis pigmentosával. Hallókészüléket 3 éves kora óta visel, első szemészeti tünete 18 éves korában jelentkezett farkasvakság formájában. Az Usher-szindrómát 26 éves korában diagnosztizálták, mindkét szemén szürkehályog-műtétet végeztek műlencse-beültetéssel öt évvel ezelőtt. Általános anamnézise eseménytelen, alapbetegsége nincs. Mindkét szeme kismértékben myop, bal oldalt astigmiával (–3,0 Dsph/–2,5 Dsph, –1,0 Dcyl 20°), két évvel ezelőtt legjobb korrigált visusa mindkét oldalon kml volt. Már húsz évvel ezelőtti látótérvizsgálatán is 5°-os csőlátás ábrázolódott. „Full-field” ERG-vizsgálatán a scotopikus és a fotopikus válasz megszűnt; multifokális ERG-n súlyosan csökkent amplitúdók a centrális gyűrűkben. Fundusán réslámpás vizsgálattal halvány papilla, szűkebb erek, atrófiás makulatáj, a periférián csontsejtszerű és paravasalis pigmentáció, illetve nummuláris pigmentkicsapódás látható. Fundusfotó, FAF- és OCT-vizsgálat is készült (5-6. ábrák). Genetikai vizsgálatának eredménye heterozigóta kereteltolódást okozó duplikációt és egy aminosavcserét eredményező pontmutációt mutatott az USH2A génben (1. táblázat).
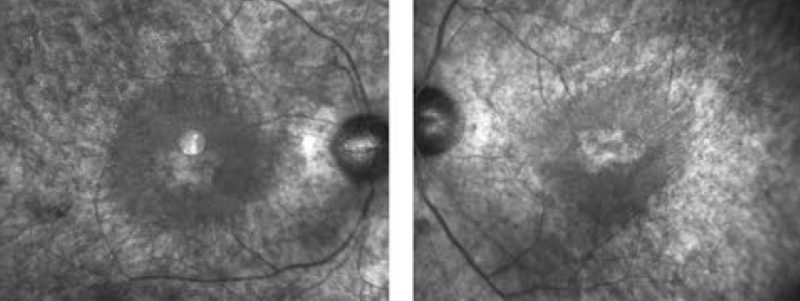
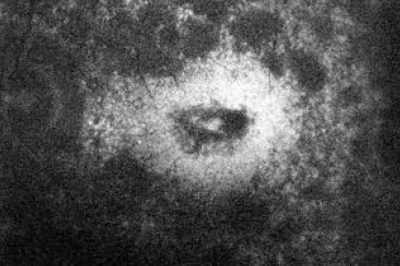
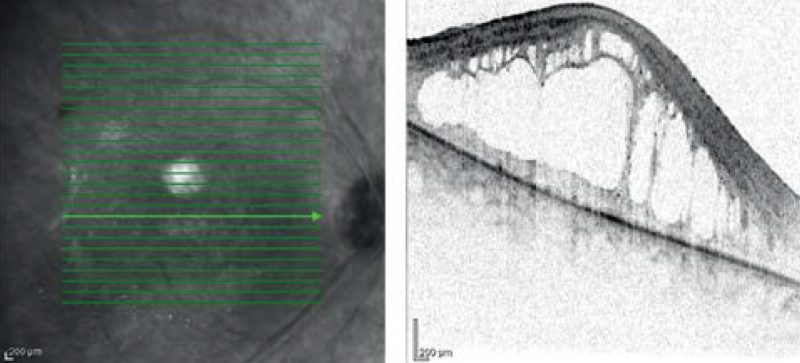
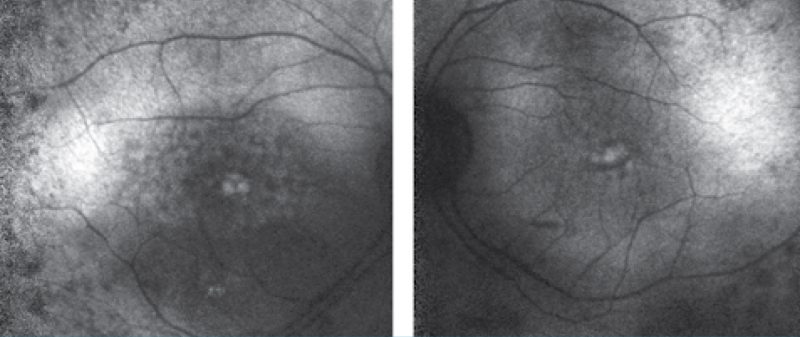
Negyedik betegünk egy 43 éves nő, aki szintén szindrómás retinitis pigmentosával fordult hozzánk. Hallássérültségére 4 éves korában derült fény, hallókészüléket 5 éves korában kapott. Farkasvaksága 20 éves korában jelentkezett, az Usher- szindrómát 30 éves korában diagnosztizálták. Családi anamnézisében apai ágon myopia és időskori makuladegeneráció szerepel. Első vizsgálatakor legjobb korrigált visusa mindkét oldalt 0,1, amely azóta enyhe romlást mutat; látótere 10 és 20° közötti tartományra szűkült be. „Full-field” ERG-vizsgálatán a scotopikus és a fotopikus válasz megszűnt; multifokális ERG-n súlyosan csökkent amplitúdók a centrális gyűrűkben. Fundusán réslámpás vizsgálattal halvány papilla, szűkebb erek, makulatájon ödéma, periférián csontsejtszerű pigmentáció és a középperiférián pigmentkicsapódás volt látható. FAF- és OCT-vizsgálat is készült (7–8. ábrák). Genetikai vizsgálatának eredménye heterozigóta pontmutációt és egy bázispárnyi deléciót mutatott az USH2A génben (1. táblázat).
Eredményeinket az 1. táblázatban összegezzük.
Következtetés
A részletes klinikai, képalkotó és elektrofiziológiai vizsgálatokkal, illetve a genotipizálás elvégzésével mind a fenotípus, mind a genotípus dokumentálása megtörtént.
Eseteink elemzése azt mutatta, hogy a súlyos fenotípus hátterében, mind a négy esetben az USH2A gén DNS-szekvenciahosszának megváltozásával járó mutációk állnak, duplikáció esetén bázispár-többlet, deléció esetén bázispár-hiány magyarázza a fehérjefunkció kifejezett károsodását. Misszensz-mutációk esetén a DNS-lánc hossza nem változik, hanem a nukleotidcsere eredményeképp aminosavcsere történik, amely a harmadlagos fehérjeszerkezetben okoz jelentős funkcióváltozást eredményező eltéréseket.
A szemészeti genetikai betegségek több szervrendszer funkciójának a károsodásával is járhatnak, ezért fontos a betegek felismerése és megfelelő vizsgálata. Alapos gyanú esetén, például az Usher-szindrómában szimultán látás- és hallásromlás mellett célszerű a pácienst szemészeti genetikával foglalkozó centrumba irányítani. Ez azért elengedhetetlen, mert ez a betegségcsoport progresszív és jelentős életminőség-romlást okoz, amelyre a betegnek fel kell készülnie. A szemészeti- és a genetikai tanácsadás segítségével a betegek tudatosan tudják tervezni jövőjüket mind életviteli, mind családtervezési szempontból.
Az utóbbi években azonban számos kutatás folyik a genetikai betegségek gyógyítására, amelyek közül a legtöbb még preklinikai fázisban tart. A módszerek között olyan génterápiás eljárások vannak, mint a víruskarrier (adeno-asszociált- és lentivírusok) segítségével történő hiányzó gén bevitele („gene replacement”), az antiszensz RNS segítségével (ASO, „antisense oligonucleotide”) történő génexpresszió-csökkentés, valamint a CRISPR-Cas9 bakteriális enzimrendszerrel történő génszerkesztés („gene editing”). Kifejezetten az USH2A gén 13. exonjának mutációs „hot spotjában” található mutációkra klinikai vizsgálat zajlik az ultevursen nevű antiszensz oligonukleotiddal, amely az említett exont kizárja az átíródásból (transzlációból) ezáltal rövidebb, azonban funkcióképes fehérje keletkezik (exon skipping). Ezek a technológiák igen ígéretesek a jövőben alkalmazható molekuláris terápiák tekintetében, azonban jelenleg még fejlesztés alatt állnak. Betegeink rendszerint tájékozottak a kutatások alakulásával kapcsolatban, de fontos számukra hangsúlyozni, hogy az ígéretes kísérleti eljárások számukra jelenleg még nem alkalmazhatók a klinikai gyakorlatban (3–5).
Nyilatkozat
A szerzők kijelentik, hogy speciális eseteket ismertető eredeti közleményük megírásával kapcsolatban nem áll fenn velük szemben pénzügyi vagy egyéb lényeges összeütközés, összeférhetetlenségi ok, amely befolyásolhatja a közleményben bemutatott eredményeket, az abból levont következtetéseket vagy azok értelmezését
Rövidítések: BCVA: legjobb korrigált visus („best corrected visual acuity”), Dsph: szférikus dioptria, ERG: elektroretinográfia, FAF: fundus autofluoreszcencia, fé: fényérzés, IR: infravörös („infrared”), kml: kézmozgás látás, RNS: ribonukleinsav, NGS: új generációs szekvenálás („new generation sequencing”), OCT: optikai koherencia tomográfia
Irodalom
1. Fuster-García C, García-Bohórquez B, Rodríguez-Muñoz A, et al. Usher Syndrome: Genetics of a Human Ciliopathy. International Journal of Molecular Sciences 2021; 22(13): 6723.
https://doi.org/10.3390/ijms22136723
2. Zhu T, Chen D, Wang L, et al. USH2A variants in Chinese patients with Usher syndrome type II and non-syndromic retinitis pigmentosa. British Journal of Ophthalmology 2021; 105: 694–703. Epub 2020 Jul 16.
https://doi.org/10.1136/bjophthalmol-2019-315786
3. Toms M, Pagarkar W, Moosajee M. Usher syndrome: clinical features, molecular genetics and advancing therapeutics. Therapeutic Advances in Ophthalmology January 2020; 12: 1–19.
https://doi.org/10.1177/2515841420952194
4. Gwenaelle GS, Géléoc Aziz El-Amraoui. Disease mechanisms and gene therapy for Usher syndrome, Hearing Research 2020; 394: 107932. ISSN 0378-5955,
https://doi.org/10.1016/j.heares.2020.107932
5. Ledford, Heidi. CRISPR treatment inserted directly into the body for first time. Nature 2020; 579(7798): 185–186.
https://doi.org/10.1038/d41586-020-00655-8