First Genetically Confirmed Case of Occult Macular Dystrophy (OMD) in Hungary (A Case Report)
doi: 10.55342/szemhungarica.2025.162.2.83
Case report
Summary
The article describes a case study of occult macular dystrophy (OMD), a rare autosomal dominant retinal disorder causing progressive central vision loss without visible retinal abnormalities. The condition’s subtle presentation often leads to misdiagnosis, as standard retinal exams typically appear normal. Diagnosis relies on electrophysiological tests, particularly multifocal electroretinograms (ERG), which show reduced central retinal responses despite the normal full-field ERG results. Advances in optical coherence tomography (OCT) have enabled the identification of microstructural anomalies in the photoreceptor layers. However, the definitive diagnosis of OMD can only be confirmed through genetic sequencing, which identifies specific pathogenic variants associated with the disorder. The patient first presented with mild myopia at age 14, with normal retinal findings. Over the years, her vision progressively declined, culminating in significant central visual field loss by age 38. Optical coherence tomography showed microstructural retinal abnormalities, suggesting a possible diagnosis of occult macular dystrophy. Even though the full-field ERG results were normal, the multifocal ERG test revealed lower central responses, which match the signs of OMD. Genetic testing confirmed OMD, identifying a pathogenic RP1L1 gene variant. This study emphasises the importance of early, comprehensive diagnostics, including electrophysiological and genetic testing, for accurate OMD diagnoses. It also highlights the need for genetic counselling due to the condition’s dominant inheritance and variable penetrance. This case is the first genetically confirmed OMD reported in Hungary, underscoring the rarity of the condition and the diagnostic challenges it presents in clinical practice.
Összefoglaló
A közlemény az okkult makuladisztrófia (OMD), egy ritka, autoszomális domináns retinalis rendellenesség esetét ismerteti, amely látható retinalis eltérések nélkül progresszív centrális látásvesztést okoz. Az állapot enyhe fokú megjelenése gyakran téves diagnózishoz vezet, mivel a standard retinavizsgálatok általában negatív eredménnyel zárulnak. A diagnózis elektrofiziológiai vizsgálatokra támaszkodik, különösen a multifokális elektroretinogramra (ERG), amely a normális Ganzfeld-ERG-eredmények ellenére csökkent központi retinaválaszokat mutat. Az OMD végleges diagnózisa azonban csak genetikai szekvenálással erősíthető meg, amely azonosítja a rendellenességgel összefüggő specifikus patogén variánsokat. A közlemény egy páciens esetét írja le, aki először 14 éves korában jelentkezett kisfokú rövidlátással, negatív retinaleletekkel. Az évek során látása fokozatosan romlott, ami 38 éves korára jelentős centrális látótérveszteséghez vezetett. Az optikai koherencia tomográfia (OCT) mikroszerkezeti retinarendellenességeket jelzett, ami az okkult makuladisztrófia (OMD) gyanúját vetette fel. Az elektrofiziológiai vizsgálat csökkent centrális válaszokat mutatott ki a multifokális ERG-n, ami összhangban volt az OMD-vel, a normális Ganzfeld-ERG-eredmények ellenére. A genetikai vizsgálat megerősítette az OMD diagnózist, azonosítva a patogén RP1L1 génváltozatot. Ez az eset hangsúlyozza a korai, átfogó diagnosztika fontosságát, beleértve az elektrofiziológiai vizsgálatokat is a pontos OMD-diagnózis érdekében. Kiemeljük a genetikai tanácsadás szükségességét az állapot domináns öröklődése és változó penetrációja miatt. Ez az eset az első genetikailag igazolt OMD Magyarországon, kiemeljük a betegség ritkaságát és a klinikai gyakorlatban jelentkező diagnosztikai kihívásokat.Keywords
occult macular dystrophy (OMD), genetic sequencing, electrophysiology, (ERG), RP1L1 gene variant, central vision loss
Kulcsszavak
okkult makuláris disztrófia (OMD), genetikai szekvenálás, elektrofiziológia (ERG), RP1L1 gén variáns, centrális látásvesztés
Bevezetés
Az okkult makuladisztrófia (OMD) a klinikusok számára diagnosztikai nehézséget jelenthet nehezen megfogható jellege és megjelenési formája miatt. Előfordulása demográfiai régiónként eltérő lehet, hiszen míg az Egyesült Államokban vagy Európában ritka betegségnek számít, addig a kelet-ázsiai populációban a leggyakoribb makuladisztrófia. Az OMD egy autoszomális domináns öröklődésű kórkép, klinikailag jellemző a centrális látóélesség progresszív romlása, látható szemfenéki eltérések nélkül. A diagnózis főként a fokális vagy a multifokális elektroretinogram (ERG) csökkent válaszain alapul, míg a Ganzfeld-ERG normális válaszokat mutat (5). Az optikai koherencia tomográfia (OCT) technológia elterjedésével és fejlődésével az elektrofiziológiai módszerekkel igazolt OMD-s esetekben strukturális rendellenességeket találtak az oftalmoszkóposan normál megjelenésű makulákban. Az SD-OCT képalkotás finom morfológiai változásokat mutatott ki, beleértve a fotoreceptor-réteg ellipszoid zónájának (EZ) elmosódását vagy a csapok külső szegmenseinek hiányát (8).
Az észrevehető szemfenéki elváltozások hiánya miatt a betegség diagnosztizálása nehéz lehet, jellemzi a változó penetrancia egy családon belül, valamint a betegség kialakulásának életkori különbözőségei is. Az OMD-vel foglalkozó kutatások nagy része Kelet-Ázsiából származik, de nem világos, hogy ez a fokozott tudatosságnak és a szűrési protokolloknak vagy a megnövekedett előfordulási gyakoriságnak köszönhető-e (4).
A bemutatott eset rávilágít az OCT, az elektrofiziológiai és a genetikai vizsgálatok fontosságára az OMD diagnosztizálásában.
Esetismertetés
1999-ben, rövidlátás miatt szemüveget viselő, lovas sportot űző, 14 éves nőbeteg, kontaktlencse-igénnyel kereste fel klinikánk ezzel foglalkozó rendelését. Szemészeti státuszából kiemelendő, hogy szemüvegét rendszertelenül viselte. Korrigált látóélessége jobb szemen: –2,5 D sph 0,9, bal szemen: –3,0 D sph 0,9. Szemfenék mindkét oldalon ép volt. Ekkor lágy típusú kontaktlencsét kapott, amelyet éveken át panaszmentesen viselt.
2012-ben látásromlás miatt ismét vizsgálatra érkezett, ekkor a látóélessége jobb szemen –3,0 D sph 0,6, bal szemen –3,75 D sph 0,5 volt. Látótere teljes, de fokozódó látásromlást élt meg, a beeső fények extrém módon zavarták. Visusvizsgálatkor a különböző megvilágítású táblákon különböző látóélességet jelzett.
2018-ban elvégzett vizsgálatakor Goldmann-periméterrel relatív centrális scotomát regisztráltak, jobb<bal, a kritikus fúziós frekvencia (CFF) jelentős eltérést nem mutatott (40/38 Hz). Látóélessége a hátulról megvilágított olvasótáblán vizsgálva 0,3, a klasszikus olvasótábla használatával 0,5 volt mindkét szemen. Szemfenekén pupillatágítás mellett elváltozást továbbra sem észleltünk. A látóélesség csökkenésének tisztázására neurológiai vizsgálat, koponyaképalkotó-vizsgálat (MR), illetve laboratóriumi vizsgálatok sora következett, de a kivizsgálások érdemi eltérést nem igazoltak.
2023-ban a legjobb korrigált visus értéke már csak 0,1 mindkét oldalon. Centrális látótér-defektusai fokozódtak, a külső határok épsége mellett. A makula-OCT képein mikrostrukturális elváltozások, a fotoreceptorok külső szegmenseinek egyenetlensége, rövidülése mutatkozott. Az előzetes kórtörténet, a mikroperimetria során mutatkozó relatív centrális defektusok és az OCT-képeken észlelt diszkrét elváltozások következtében felmerült az okkult makuladisztrófia (OMD) diagnózisa.
Az OMD diagnózisát végül elektrofiziológiai és genetikai vizsgálatokkal sikerült igazolni. A kórképre jellemzően, nőbetegünk esetében is, normál Ganzfeld-ERG-válaszok ellenére a multifokális ERG-n csökkent centrális válaszokat regisztráltunk (1. ábra). A diagnosztikus célú genotipizálás, amely új generációs szekvenálással történt (Retinal Dystrophy NGS Panel), az RP1L1 gén c.133C>T, p.(Arg45Trp) heterozigóta missense patogén variánsát azonosította.
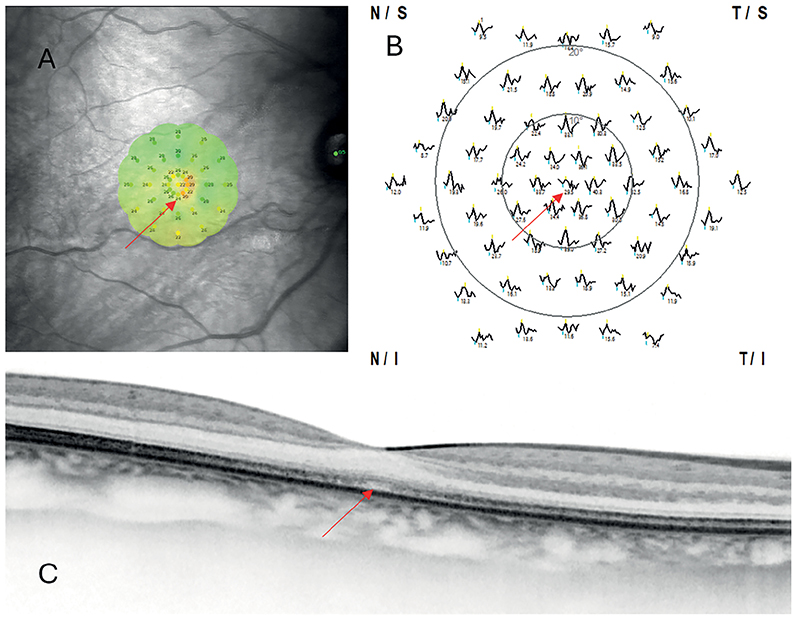
Megbeszélés
Az örökletes retinadisztrófiák a fotoreceptorokat és az RPE-t érintő ritka betegségek heterogén csoportja. Az OMD-t a szakirodalomban általában autoszomális domináns betegségként írják le, amelyet a retinitis pigmentosa 1-like 1 (RP1L1) gén mutációja okoz. Az RP1L1 gén megtalálható mind a pálcikákban, mind a csapokban. Pontos funkciója továbbra sem ismert, de az érintett betegek strukturális és elektrofiziológiai jellemzői alapján azt feltételezik, hogy a kódolt fehérje a külső fotoreceptor-szegmensek működésében vesz részt (1, 7).
2010-ben Akahori és munkatársai összefüggést írtak le az OMD és az RP1L1 gén között. Azóta számos OMD-esetről számol be az irodalom (1). Sokáig úgy tűnt, hogy az RP1L1 az egyetlen gén, amely autoszomális domináns OMD-t okoz, de a legújabb, elsősorban kelet-ázsiai betegcsoportokon végzett kutatások alapján felmerül a CRX és a GUCY2D gének kóroki szerepe is, amelyekről ismert, hogy autoszomális domináns csapdisztrófiát okozhatnak (4, 6, 9). Fujinami és munkatársai ajánlása szerint az OMD három alkategóriára osztható: RP1L1-asszociált OMD (Miyake-kór), egyéb örökletes OMD, amelyet más génvariánsok okoznak, és nem örökletes okkult makuladisztrófia-szerű szindróma (progresszív okkult makulopathia). Ez a nomenklatúra szükségesnek tűnt, mivel a morfológiai fenotípusok és a genotípusok között jelentős összefüggés mutatható ki, ami az OMD hátterében álló különböző patofiziológiai folyamatokra utal (3, 4). Az RP1L1 génben az ázsiai és az európai populációban is a leggyakoribb a heterozigóta c.133C>T, p.(Arg45Trp) patogén variáns.
Betegünk esetében is ez a genetikai eltérés igazolódott.
Zobor és munkatársai 2018-ban leírták, hogy egy nagy, főként német kohorszban is minden beteg a klasszikus leleteket mutatta, beleértve a csökkent korrigált visust, a centrális látótérdefektusokat, a kóros multifokális ERG-t, normális fundusképet, a fotoreceptorok mikrostruktúrájának SD-OCT-vel detektált tipikus elváltozásai mellett. Az életkor, amelyben a betegek látásromlást észlelnek, jelentősen eltér. A látásélesség folyamatos csökkenésének időtartama 10 és 30 év között változott a tanulmányok szerint (7, 8).
Eset jelentősége
Legjobb tudomásunk szerint Magyarországon ez az első publikált OMD-eset, amelyet genetikailag igazoltan az RP1L1 gén patogén variánsa okoz. Ez alátámasztja, hogy mennyire ritka és gyakran fel nem ismert ez a betegség a mindennapi klinikai gyakorlatban. Az OMD-t más retinadegenerációk differenciáldiagnózisaként mindenképpen figyelembe kell vennünk a normálisnak tűnő szemfenéki képpel és egyébként megmagyarázhatatlan látóélesség-romlással jelentkező betegeknél. Az elektrofiziológiára és részletes funkcióvizsgálatra támaszkodó szemészeti vizsgálat és a retina molekuláris genetikájával foglalkozó tapasztalt laboratóriummal való együttműködés lehetővé teszi az OMD diagnózisának felállítását. Ez megkímélheti a beteget a neurológiai és képalkotó vizsgálatok sokaságától, és ezeknek jelentős anyagi terhétől. Fontos kiemelni a domináns öröklésmenet és a változó penetrancia miatt a genetikai tanácsadás jelentőségét, hiszen a családtagok érintettségével is számolnunk kell.
Nyilatkozat
A szerzők kijelentik, hogy az esetismertetés megírásával kapcsolatban nem áll fenn velük szemben pénzügyi vagy egyéb lényeges összeütközés, összeférhetetlenségi ok, amely befolyásolhatja a közleményben bemutatott eredményeket, az abból levont következtetéseket vagy azok értelmezését.
Irodalom
1. Akahori M, Tsunoda K, Miyake Y, Fukuda Y, Ishiura H, Tsuji S, et al. Dominant mutations in RP1L1 are responsible for occult macular dystrophy. Am J Hum Genet 2010; 87(3): 424–9.
https://doi.org/10.1016/j.ajhg.2010.08.009
2. Conte I, Lestingi M, den Hollander A, Alfano G, Ziviello C, Pugliese M, et al. Identification and characterisation of the retinitis pigmentosa 1-like1 gene (RP1L1): a novel candidate for retinal degenerations. Eur J Hum Genet 2003; 11(2): 155–62.
https://doi.org/10.1038/sj.ejhg.5200942
3. Fujinami K, Kameya S, Kikuchi S, Ueno S, Kondo M, Hayashi T, et al. Novel RP1L1 Variants and Genotype-Photoreceptor Microstructural Phenotype Associations in Cohort of Japanese Patients With Occult Macular Dystrophy. Invest Ophthalmol Vis Sci 2016; 57(11): 4837–46.
https://doi.org/10.1167/iovs.16-19670
4. Fujinami-Yokokawa Y, Yang L, Joo K, Tsunoda K, Liu X, Kondo M, et al. Occult Macular Dysfunction Syndrome: Identification of Multiple Pathologies in a Clinical Spectrum of Macular Dysfunction with Normal Fundus in East Asian Patients: EAOMD Report No. 5. Genes (Basel) 2023; 14(10).
https://doi.org/10.3390/genes14101869https://doi.org/10.3390/genes14101869
5. Kabuto T, Takahashi H, Goto-Fukuura Y, Igarashi T, Akahori M, Kameya S, et al. A new mutation in the RP1L1 gene in a patient with occult macular dystrophy associated with a depolarizing pattern of focal macular electroretinograms. Mol Vis 2012; 18: 1031–9. PMID: 22605915 PMCID: PMC3351429
6. Kitiratschky VB, Nagy D, Zabel T, Zrenner E, Wissinger B, Kohl S, et al. Cone and cone-rod dystrophy segregating in the same pedigree due to the same novel CRX gene mutation. Br J Ophthalmol 2008; 92(8): 1086–91.
https://doi.org/10.1136/bjo.2007.133231
7. Miyake Y, Tsunoda K. Occult macular dystrophy. Jpn J Ophthalmol 2015; 59(2): 71–80.
https://doi.org/10.1007/s10384-015-0371-7
8. Zobor D, Zobor G, Hipp S, Baumann B, Weisschuh N, Biskup S, et al. Phenotype Variations Caused by Mutations in the RP1L1 Gene in a Large Mainly German Cohort. Invest Ophthalmol Vis Sci 2018; 59(7): 3041–52.
https://doi.org/10.1167/iovs.18-24033
9. Zobor D, Zrenner E, Wissinger B, Kohl S, Jagle H. GUCY2D- or GUCA1A-related autosomal dominant cone-rod dystrophy: is there a phenotypic difference? Retina 2014; 34(8): 1576–87.
https://doi.org/10.1097/IAE.0000000000000129