Current status of genetic therapies in Ophthalmology
doi: 10.55342/szemhungarica.2023.160.4.154
Review
Summary
In this paper, as an update to the publication “Genetics in Ophthalmology” (published in Szemészet in 2014), the authors would like to summarize the developments in ophthalmic genetics: new results in biotechnology, diagnostics and therapy. The topmost breakthrough in recent years has been the registration and introduction into clinical practice of voretigene-neparvovec, a drug developed for the treatment of Leber congenital amaurosis (LCA) and retinitis pigmentosa (RP) caused by the mutations of the RPE65 gene. In July 2022, the Department of Ophthalmology at Semmelweis University received accreditation as an Ophthalmic Gene Therapy Centre and since then 10 treatments for patients with LCA have been performed. In addition to the treatments, there are many other research and development projects in gene therapy for inherited and acquired diseases. Along with the therapy, genetic diagnostics has also undergone significant development in recent years, with methods becoming significantly faster and more cost-efficient, making them more widely available. In the last one and a half years, more than 500 Hungarian patients with hereditary retinal dystrophy have been genotyped in Hungary in the framework of research collaborations.
Összefoglaló
A Szemészet hasábjain 2014-ben megjelent Genetika a szemészetben c. továbbképző közlemény folytatásaként jelen munkánkban szeretnénk összefoglalni a szemészeti genetika területén azóta elért biotechnológiai, diagnosztikus és terápiás eredményeket. Az elmúlt évek legnagyobb áttörése egyértelműen az RPE65-gén hibái okozta Leber congenitalis amaurosis (LCA) és retinitis pigmentosa (RP) kezelésére kifejlesztett voretigene-neparvovec törzskönyvezése és bevezetése a klinikai gyakorlatba. 2022 júliusában a Semmelweis Egyetem Szemészeti Klinikája akkreditációt kapott, mint Szemészeti Génterápiás Centrum és azóta megtörtént az első 10 kezelés LCA-ban szenvedő betegekben. A már bevezetett kezelés mellett számos más génterápiás kutatás, fejlesztés történik mind öröklődő, mind szerzett betegségek esetében. A terápiával párhuzamosan a genetikai diagnosztika is jelentős fejlődésen ment keresztül az elmúlt években: a módszerek jelentősen gyorsabbak és költséghatékonyabbak, ezáltal minden eddiginél szélesebb körben elérhetőek lettek. Kutatási együttműködések keretében hazánkban az elmúlt másfél évben több, mint 500 magyar öröklődő retinadisztrófiás beteg genotipizálása történt meg.Keywords
ophthalmic genetics, gene therapy, inherited retinal dystrophies, genetic counselling
Kulcsszavak
szemészeti genetika, génterápia, öröklődő retinadisztrófiák, genetikai tanácsadás
Bevezető
A Szemészet hasábjain Genetika a szemészetben c. továbbképző közleményben 2014-ben összefoglaltuk a szemészeti genetikai akkori aktuális állapotát. Jelen közleményben hivatkozunk az abban leírt alapfogalmakra (1).
A molekuláris genetika és a biotechnológia fejlődése új lehetőségeket hozott az utóbbi tíz évben az öröklődő retinabetegséggel élő páciensek diagnosztikájában, követésében és lehetséges gyógyításában.
A nemzetközi Human Genom Project, amelynek célja a teljes humán genom szekvenálása, azaz DNS-lánc minden betűjének a meghatározása volt, hivatalosan 1990-ben kezdődött. Ez a vállalkozás az akkor elérhető technológiával rengeteg kihívást rejtett és rendkívül idő- és munkaigényes folyamat volt. A projekt 2003-ban publikálta az első referenciaszekvenciát az emberi génállomány 92%-ának feltérképezésével. A hiányzó szakaszok teljes felfedését csupán 2022 januárjában fejezték be. Az emberi genom kifeszítve 1,8 méter hosszú lenne, a 23 kromoszómán elhelyezkedve 3,2 milliárd bázispárt (betűt) tesz ki és több mint 23 000 gént tartalmaz (2, 3).
Az emberi génállomány szabadon elérhető adatbázisokban való publikálása óriási lendületet adott a molekuláris genetikai vizsgálatoknak. Exponenciálisan nőtt a betegségeket okozó beazonosított gének száma, gén-gén interakciókat írtak le, felfedeztek számos génexpressziót szabályozó folyamatot, jelátviteli útvonalakat, illetve megismertük az epigenetikus szabályozást (3).
A genom fehérjéket kódoló szakaszait exonnak, a közöttük elhelyezkedő nem kódoló régiókat intronnak nevezzük. Az emberi genom körülbelül 180 000 exont tartalmaz, amelyeket együttesen exomnak nevezünk. Az exom az emberi genom körülbelül 1%-át teszi ki, és így körülbelül 30 millió nukleotidot tartalmaz (3).
A gén egy adott fehérjét (fehérjealegységet) kódoló DNS-szakasz, amely általában több exonból és a köztük elhelyezkedő intronból áll. Az intronok a teljes genom kb. 98%-át teszik ki és mára már biztos, hogy nem „haszontalan (junk) DNS”, hanem fontos szabályozó szerepük van. Egyre több, az intronokban elhelyezkedő (deep intronic), betegséget okozó variáns kerül publikálásra (4, 5, 6). Mivel ezen elváltozások strukturális hatása nem egyértelmű, sejttenyészeteken történő funkcionális vizsgálatok segítenek bizonyítani a szabályozásban játszott szerepüket. A nagy géneket (pl. 50 exon feletti méretűeket) nehezebb szekvenálni a klasszikus Sanger-féle szekvenálással, illetve kihívást jelent és gyakran nem is lehetséges a vírusvektorba való becsomagolásuk.
Manapság az újgenerációs teljes exomszekvenálás (whole exome sequencing = WES) módszere terjedt el, mivel általa a klinikailag releváns gének exonjait idő és költséghatékonyan lehet egy futtatás alatt párhuzamosan vizsgálni (massive paralell sequencing). A teljes exomszekvenálás költsége a piacra kerülő modern újgenerációs szekvenáló készülékek kapacitásnövekedésével az utóbbi években jelentősen csökkent. A szekvenálás során a legnehezebb kihívás az eredmények értelmezése, a talált variánsok interpretációja. Minden egyes exomszekvenálás során körülbelül 120 000 eltérést detektálhatunk a referenciaszekvenciától, amelyeket a megfelelő bioinformatikai algoritmussal szűrve találhatjuk meg a vizsgált betegséggel valóban összefüggésben álló eltéréseket (7). Ezeket az eltéréseket korábban gyakoriságuk és patogenitásuk alapján polimorfizmusoknak, illetve mutációknak hívtuk, ma egységesen variánsnak nevezzük őket. Az American College of Medical Genetics and Genomics (ACMG) kezdeményezésével kialakítottak egy egységes, 5 kategóriából álló klasszifikáló rendszert, amelyben a mendeli öröklődés menetű kórképeket és azokhoz köthető genetikai variánsokat osztályozták. Az 5 csoport a következőkből áll:
- patogén,
- valószínűleg patogén,
- ismeretlen jelentőségű (VUS – variant of unknown significance),
- valószínűleg jóindulatú és
- jóindulatú variáns.
Az utolsó esetén jelen tudásunk szerint nem áll fenn összefüggés a gén és a betegség között, az első esetében egyértelműen látható asszociáció a genotípus és fenotípus között. A VUS esetében nincs elegendő adat a genetikai vizsgálat eredménye és a betegség közötti kapcsolat felállításához (8). A WES-eredmények megfelelő értelmezéséhez elengedhetetlen az adott gének öröklődésmenetének és penetranciájának, illetve a genetika és az orvostudomány adott területének mély ismerete (pl. kardiogenetika, oftalmogenetika, nefrogenetika, mitokondriális genetika stb.) (9, 10).
Genetikai tanácsadás a szemészetben
Az öröklődő retinadisztrófiák csoportja klinikai és genetikai szempontból rendkívül heterogén. A betegek kivizsgálása során alapos szemészeti és multimodális képalkotó vizsgálatot (Heidelberg Spectralis OCT, infravörös, kék autofluoreszcencia), családfaelemzést, színlátás (Ishihara-tábla, Farnsworth-teszt, Cambridge Color Test), látótér (Goldmann kinetikus vagy automata perimetria), illetve elektrofiziológiai vizsgálatokat végzünk (Ganzfeld és multifokális ERG, EOG, VEP, kromatikus pupillometria, sötétadaptometria). A nemzetközi irodalomban, kutatásokban ezt nevezik deep phenotypingnak.
A Semmelweis Egyetem Szemészeti Klinikáján több mint 30 éve működik Elektrofiziológiai labor, amelynek működése során számos kutatás valósult meg az öröklődő retinadisztróiák területén. A nagylétszámú (közel ezer fő) betegcsoportunk diagnosztikája és gondozása összetett, rendkívül idő- és eszközigényes feladat. A Szemészeti Genetika szakrendelés 2019-ben kezdte meg működését a Szemészeti Klinikán, fő profilja az öröklődő retinabetegek ellátása, de ezenkívül a szem egyéb öröklődő kórképeivel, például a látóideg öröklődő betegségeivel, mitokondriális betegségekkel, valamint egyéb szindrómás betegségekkel küzdő szemészeti betegekkel is foglalkozik.
A molekuláris genetika fentebb leírt lendületes fejlődése megkönnyíti a genetikai diagnosztikát a szemészeti genetika területén is. A genetikai heterogenitás miatt az újgenerációs célzott panelek közül a szemészetben a nagy paneleket alkalmazzuk, amelyek több mint 350 gént tartalmaznak és a kópiaszám-változásokat (nagyobb deléciókat és inszerciókat) is felderítik az újgenerációs szekvenálás módszerével. A szemészeti genetikai vizsgálatot preteszt (teszt előtti) genetikai tanácsadás előzi meg. Ennek során a szemészeti genetikával foglalkozó szakember tájékoztatja a beteget a genetikai vizsgálat módszeréről, a genetikai információ jelentéséről, nyilatkoztatja a beteget az incidentális találatok közléséről.
A genetikai vizsgálat eredményét posztteszt (teszt utáni) genetikai tanácsadáson beszéljük meg a beteggel. A genetikai vizsgálat sikeraránya (vagyis amikor egyértelműen azonosítható a patogén genetikai eltérés) nemzetközi közlések alapján szemészeti betegségek esetén kb. 60-80%. Azonban tájékoztatni kell a beteget arról is, hogy a genetikai vizsgálata negatív eredménnyel is zárulhat, és akár hosszú évekig elhúzódhat. A genetikai vizsgálatot jellemzően certifikált molekuláris genetikai laboratóriumok végzik diagnosztikai és/vagy kutatási célból. A diagnosztikai vizsgálatokat jellemzően az ellátórendszer, a kutatási vizsgálatokat a kutató intézet finanszírozza. Ez utóbbi esetben az eredményre azonban gyakran hosszabb ideig (akár több évig) is várni kell. A beteggel kapcsolatos genetikai információ és lelet a páciens tulajdona, a vizsgálat eredménye a többi egészségügyi adathoz hasonlóan titkos, szenzitív adat.
Miért lehet hasznos a genetikai vizsgálat?
A genetikai vizsgálat lerövidítheti a pontos diagnózis felállításának időtartamát, ezáltal megkímélve a beteget és hozzátartozóit hosszú évek bizonytalanságától. A genetikai vizsgálat eredményét mindig klinikai szempontból kell értelmeznünk. Fontos tudni, hogy a negatív eredmény nem jelenti a mutáció és a betegség nemlétét, hanem hogy máshol és más módszerrel kell tovább keresnünk a választ. Hasonlóan, a pozitív eredmény nem mindig jelenti a variáns betegséget okozó voltát, ezért a családtagokat is meg kell vizsgálnunk genetikai szempontból. Gyakran előfordul, hogy bizonytalan jelentőségű variánst ír le a genetikai lelet, amely későbbi újraértékelést/revalidálást igényel. Habár számos összefüggést azonosítottak a genetikai mutációk és a betegségek megjelenése között, ez a kapcsolat nem mindig egyértelmű, mivel környezeti, genetikai és egyéb módosító tényezők is befolyásolják. Akár azonos családból származó és azonos genetikai mutációval bíró páciensek esetében is eltérő lehet a manifesztálódó klinikai kép, emiatt óvatosan kell fogalmaznunk a betegség hosszú távú progresszióját illetően. A diagnózis felállításakor a betegben és hozzátartozóiban számos kérdés merül fel a látásával kapcsolatban. A genetikai diagnózis segíthet, hogy a páciens személyes döntéseket hozhasson a továbbtanulása, a munkavállalása, az életvezetése és a családtervezése terén. Gyermekek számára feltétlen javasolt a látásvizsgáló bizottság felkeresése, amely fővárosi és országos illetékességű szakértői bizottsági tevékenységet végez funkcionális látásfelméréssel, szakértői véleményt ad a pedagógus számára, pontosan meghatározva, hogy a látássérült gyermeket milyen mentességek, könnyítések, illetve többletidő illeti meg a tanulmányai és vizsgái során (https://fpsz.hu/fpsz-latasvizsgalo-gyogypedagogiai-tanacsado-korai-fejleszto-oktato-es-gondozo-tagintezmenye/). A felnőttek számára a Vakok és Gyengénlátók Szövetsége, illetve elemi rehabilitációval foglalkozó alapítványok nyújtanak többféle elérhető szolgáltatást országszerte. A fehérbotos járástanítás, biztonságos közlekedés, házimunka elvégzése, pszichológiai tanácsadás, egyéni és csoportos terápiák, informatikai képzés, segédeszközök kipróbálása és kölcsönzése, szociális segítség mind szerepel a szolgáltatásaik között számos egyéb modullal együtt (ld. Szemészet korábbi lapszáma, 11).
Amennyiben a preteszt genetikai tanácsadás keretében a beteg vagy gyámja nyilatkozott, hogy kéri az incidentális/másodlagos találatok közlését, a páciens felvilágosítást kap a genetikai vizsgálat során felfedett másodlagos variánsokról is, amelyek a jövőben orvosi beavatkozást/vizsgálatot igényelnek potenciálisan fellépő egészségügyi problémák miatt, pl. pajzsmirigy UH-vizsgálatra küldhetjük a beteget, ha esetében pajzsmirigy-karcinómára jellemző genetikai variánst fedeznek fel. Ezen másodlagos találatok ritkán, kb. az esetek 1-2%-ában fordulnak elő (12).
A genetikai vizsgálatok során családi variánselemzésre, szegregációs és hordozósági vizsgálatra is szükség lehet. Az inkomplett penetrancia és a gének eltérő expresszivitása (kifejeződése) függvényében azonban a családtagok esetén azonos variánsokat hordozva is eltérő lehet a klinikai manifesztáció. Szerepe van a környezeti hatásoknak és az egyéb genetikai módosító tényezőknek. Sajnos retinadisztrófiákban jelenleg nem ismertek megelőző, illetve a folyamatot lassító kezelések, fontos azonban hangsúlyozni a fényvédelmet, a toxikus hatások, különösen a dohányzás kerülését.
A genetikai diagnózis felállítása és a családi hordozósági vizsgálatok során gyakran felmerül a szülőkben, hogy az egészséges gyermek genetikai vizsgálatát is elvégeztessék, azonban a tünetmentes gyermekek genetikai vizsgálatát egyértelműen tiltja a magyar Humángenetikai törvény (13).
Génterápiák: génpótló kezelések, géncsendesítés, optogenetika, génszerkesztés (Gene replacement therapy, gene silencing, optogenetics, gene editing)
A génterápia gyűjtőfogalom, amelybe a rekombináns vírusvektorokat alkalmazó génpótló kezelések, a DNS-, illetve RNS-alapú genom-, illetve génszerkesztő technikák és egyéb módszerek, például az optogenetika is beletartoznak (1. táblázat).

Bizonyos genomszerkesztési módszerek már régóta ismertek, mint pl. a „zinc finger” -nukleázok, transzkripciós aktivátorszerű effektor nukleáz (transcription activator-like effector nuclease = TALEN), de a technika nagyfokú pontatlansága miatt klinikai jelentőségük kérdéses volt (14).
Génpótló kezelések
A génterápia fejlődésében az igazi áttörést a vírusvektorok alkalmazása hozta meg. Az elmúlt 25 évben az adenovírusok, adeno-asszociált vírusok és lentivírusok hosszas tanulmányozása során az adeno-asszociált vírusok (AAV) bizonyultak legalkalmasabbnak a humán felhasználásra (15). A vírusvektorokkal általában a hibás génkópia helyett egy jól működő génkópiát viszünk be a sejtbe, de lehetőség van a célsejtekkel egészen más fehérjék (pl. opszinok, gyógyszerek) termeltetésére is.
Az ex vivo génterápia során a betegből nyert sejteket laboratóriumi körülmények között módosítják, pl. kiméra antigénreceptorral felruházott T-lymphocyták = CAR T-sejt immunterápia limfoid leukémiákban. Ebben az esetben visszajuttatás előtt ellenőrizhető, hogy valóban megtörténtek-e a genetikai változtatások (16, 17).
Az in vivo génterápia során a betegben történik a génmódosítás, amely nagyobb mellékhatás-kockázattal jár és lényegi kérdés a szabályozhatóság. A terápiás génbevitellel járó kezelések során a hiányzó génkópiát a rekombináns vírusvektorba építve juttatjuk be a beteg sejtjeibe, főként autoszomális recesszív és X-kromoszómához kötött kórképekben (18). A domináns kórképekben, mint például a rhodopsin gén okozta autoszomális domináns retinitis pigmentosában, általában a kóros túlműködést kell leállítani, amelyre a gének csendesítését célzó rövid RNS-láncok (antisense oligonukleotidok) alkalmazása lehet talán megoldás (19).
A szem immunprivilegizáltsága révén rendkívül jó génterápiás célszerv, emiatt kutatócsoportok már közel 25 éve fejlesztenek lehetséges szemészeti génterápiákat.
A génpótló kezelések során a géneket a fotoreceptorokba, illetve a retinalis pigmenthámba lehet bejuttatni adeno-asszociált vírusvektorokkal subretinalis injekció, vagy intravitrealis injekció révén. Az AAV-vektorok előnye alacsony immunogenitásuk és biztonságos alkalmazhatóságuk. A vírusvektorokat alkalmazó génterápia hátránya, hogy mutációfüggő módszer, a DNS bizonyos részei véletlenszerűen kitörlődhetnek vagy módosulhatnak, illetve a vírusvektorok szállítási kapacitása véges. Nagy géneknél pl. ABCA4 (Stargardt kór), USH2A (Usher szindróma 2. típusa), MYO7A (Usher-szindróma1/B típusa), EYS (pálcika-csap disztrófia) problémát jelent a nagyméretű gén vírusvektorba csomagolása (15, 20, 21).
A génpótló kezelések egyik alapfeltétele, hogy a célszervben jelen legyenek azok a sejtek, amelyben a hibás gén a betegség kialakulásához vezet. Ezért nem alkalmazhatóak sikerrel a retinadisztrófiák előrehaladott stádiumában (14, 21), mivel a súlyosan destruált szövetben már nincs génátírásra alkalmas célsejt (1. ábra).
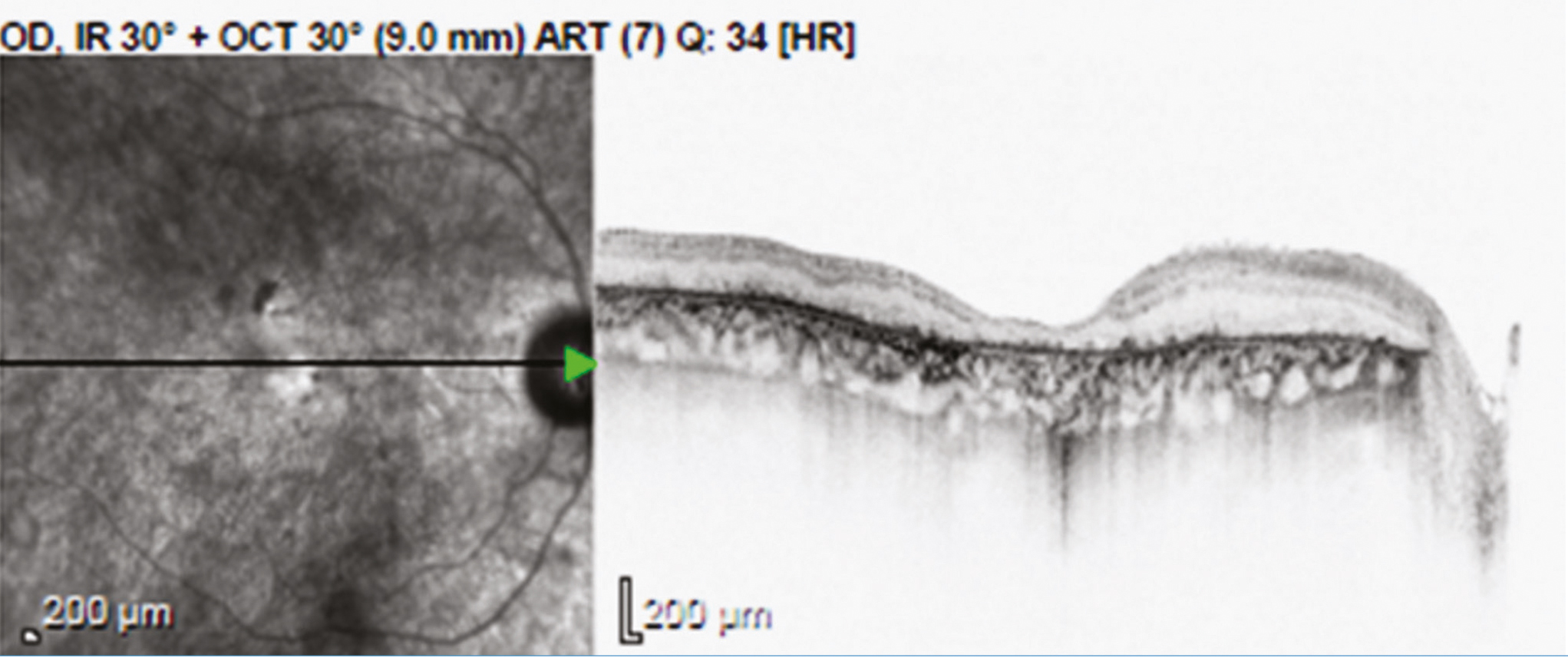
Az első törzskönyvezett génterápiás gyógyszer, amely öröklődő retinadisztrófiában alkalmazható az RPE65-gén biallélikus mutációi esetén, 2017-ben kapta meg az amerikai (FDA) és 2018-ban az európai gyógyszerészeti hatóság (EMA) engedélyét: ez a készítmény a Luxturna® (voretigene-neparvovec-rzyl), amely adeno-asszociált vírusvektorba csomagolt teljes RPE65-gént tartalmaz magas kópiaszámban. A gyógyszerből 300 ml-t szükséges altatásban végzett pars plana vitrectomia során subretinalisan beinjektálni a temporális érárkád mentén, hogy az minél hatékonyabban jusson el a célsejtekhez (RPE), és megkezdődjön a génátírás a bevitt normál génkópia alapján (22, 23).
CRISPR/Cas9 génszerkesztő technológia
A genomszerkesztésben a CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR-associated nuclease 9) technológia fejlődése hozott áttörést, s már számos állatkísérletben bizonyították, hogy pontossága révén daganatos és öröklődő betegségek gyógyítására is felhasználható.
„A CRISPR/Cas9 rendszer az egysejtűek védekezési módszere a vírusok és káros plazmidok ellen.” – írja a Wikipédia (24). Egyszerűbben fogalmazva, olyan bakteriális működési egység, amely képes a DNS-láncot vágni, így a kívánt helyen alkalmazható és a bevitt minta alapján a sejt saját DNS-javító képességét kihasználva a hibás pontot átírja a jó betűre.
Tudományosan leírva a CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR-associated nuclease 9) klaszterekben elhelyezkedő, szabályosan megszakított távolságra levő rövid (20-40 bp-nyi) palindromismétlődések között helyezkedik el a helykitöltő (spacer) DNS, amely egyedi és virális eredetű. A baktériumok kódolnak Cas-fehérjéket is, amelyek DNS-vágó enzimek. A szabályosan ismétlődő rövid palindromismétlődések, azaz CRISPR-ek, közötti helykitöltő DNS-ek alapján a Cas-fehérjék felismerik a baktériumsejtbe behatoló idegen virális nukleinsavakat és darabokra vágják őket (25, 26).
Ez a bakteriális mechanizmus több mint 30 éve ismert, elsőként 1987-ben Escherichia coliban írták le japán kutatók (26). Streptococcus pyogenesben Jennifer Doudna és Emanuelle Charpentier azonosította a CRISPR/Cas rendszert, és felismerték, hogy a rendszer alkalmas lehet RNS által programozott genomszerkesztésre. Eredményeikért 2020-ban elnyerték a kémiai Nobel-díjat (27). Emlőssejtekben Feng Zhang és kollégái alkalmazták először 2013-ban (28).
A CRISPR/Cas9, illetve CRISPR/cpf1 alkalmazása felgyorsítja a biotechnológia fejlődését, bármilyen típusú sejten alkalmazható, gyors technika (29). Az endonukleáz enzim és a specifikus vezető RNS-szakasz (guide RNA) sejtbe való bevitelével, az endonukleáz enzim a vezető RNS-szakasz által meghatározott helyen vágja el a sejt kettősszálú DNS-ét, majd a vágást követően a DNS-ből egy három nukleotidos szakaszt eltávolít, ezt követően a bevitt „mintaRNS” alapján a sejt saját DNS-javító rendszere javítja a szekvenciát, tehát egy pontmutáció, kis inszerció (nukleotidtöbblet) vagy kis deléció (nukleotidhiány) kiiktatható a DNS-láncból (27, 28).
A CRISPR/Cas9, illetve CRISPR/cpf1 rendszer előnye a gének minden korábbinál pontosabb módosítása, illetve az, hogy a kutatási termék legnagyobb része azonos maradhat, és csak a közel 20 nukleotid hosszú specifikus RNS vezető szekvenciát kell célzottan a betegséget okozó gén célszekvenciájához kifejleszteni (27, 28, 29).
A génszerkesztés előnyös az autoszomális domináns vagy szemidomináns betegségekben, amikor egy mutáns gént inaktiválnak vagy kijavítanak, például, ha egy aminosavcserét eredményező missense mutációk domináns negatív hatású vagy funkciónyerés miatt toxikus fehérje szintéziséhez vezetnek. Az öröklődő betegségek jelentős részében egy bázispárt érintő pontmutációk állnak a háttérben, ezek kijavítása a CRISPR/Cas9 rendszerrel lehetséges.
Optogenetika
Az optogenetikai eljárások technikailag hasonlóak a fent leírt génpótló kezelésekhez – a lényegi különbség, hogy ebben az esetben természetesen nem előforduló géneket juttatunk be bizonyos sejtekbe. A szemészet területén ezzel a genetikai módosítással fényérzékeny, illetve fénykibocsátásra alkalmas fehérjéket expresszáltatunk (termeltetünk) a kísérleti állatok (később a betegek) sejtjeiben, majd ezek működését egy speciális lézerrendszerrel, valamint egy ehhez csatlakozó mikroszkóppal vizsgálják és befolyásolják. A természetben, algákban és baktériumokban találhatunk fényaktivált receptorokat, fényre aktiválódó ioncsatornákat (pl. Chlamydomonas reinhardtii nevű zöldalgában, illetve egy Natronobacterium pharaonis nevű baktériumban), ezenkívül a növényekben is számos fényérzékeny fehérje-fehérje interakció figyelhető meg, amelyek segítségével a növények a fény irányát és a napszakok, évszakok változását követik (30, 31, 32, 33, 34).
Az optogenetika egy viszonylag fiatal tudományterület, kb. 20 éves múltra tekint vissza. A módszert Ernst Bamberg laboratóriumában dolgozták ki 2002-ben, amelyet Karl Deisseroth és stanfordi laborjában dolgozó kollégái, Ed Boyden és Feng Zhang, fejlesztettek tovább, amikor 2005-ben bebizonyították, hogy a csatornarodopszin segítségével tényleg aktiválható egy idegsejt (30, 31, 32, 33, 34). A Nature folyóirat az optogenetikát 2010-ben „Az év módszerévé” választotta, majd 2013-ban optogenetikusok kapták a 2013. évi idegtudományi Brain Prize-t. A dániai Grete Lundbeck Európai Agykutatási Alapítvány által létrehozott díjat megosztva kapta Ernst Bamberg, Peter Hegemann és Georg Nagel, a frankfurti Max Planck Intézet kutatói, Gero Miesenböck, a Yale Egyetemen dolgozó osztrák idegkutató, Ed Boyden, a Massachussettsi Műszaki Egyetem (MIT) kutatója és Karl Deisseroth, a Stanford Egyetem professzora. A legkorábbi kísérletekben férgek és gyümölcslegyek mozgását szabályozták fénnyel (32, 33, 34). A két legelterjedtebb fényaktiválható ioncsatorna, a pozitív ionokat átengedő csatornarodopszin (ChR), illetve a negatív ionokat átengedő kloridcsatorna, a halorodopszin (NpHR). Más-más hullámhosszú fény aktiválja őket, aminek következtében kiegészítik egymást: egyetlen sejtben párhuzamosan kifejezve lehetővé teszik, hogy ki-be lehessen kapcsolgatni az adott idegsejt aktivitását.
Egy másik új lehetőség, amelyet az optogenetika kínál, a gének transzkripciójának szabályozása igen nagy tér- és időbeli felbontással, akár több gén egyidejű kifejeződésének módosításával is. Optogenetikai kutatások révén gyógyszer hatóanyagokat vagy új, mutációfüggetlen terápiákat lehet tervezni. Számos optogenetikát alkalmazó preklinikai tanulmányt folytattak rágcsáló-, kutya- és majommodelleken, amelyben adeno-asszociált vírusvektorokba építették a megfelelő opszingéneket.
A retina több mint 60 típusú sejtet tartalmaz (35), a különböző sejtpopulációkban különböző mikrobiális, illetve állati eredetű opszinfehérjéket lehet termeltetni (expresszáltatni).
A kutatások során targetként az egész retinát, vagy a fotoreceptorokat, a bipoláris sejteket és a ganglionsejteket alkalmazták. A bejuttatás módja intravitreális injekció, illetve pars plana vitrectomia közbeni subretinalis injekció volt. Az optogenetikai fejlesztés során lényeges, milyen tulajdonságúak az opszinfehérjék – milyen a csatornák konduktivitása, ionszelektivitása, kinetikája, fény és spektrális érzékenysége, a deszenzitizációjának és regenerációjának ideje. A speciális sejttípusok optogenetikai aktiválással és csendesítéssel fényérzékennyé tehetőek, ezáltal a páciensek is vizuális érzékelést tapasztalhatnak meg (36, 37, 38).
Roska Botond és kutatócsoportja egyik fő kutatási területe a sejttípus-specifikus optogenetikai fényérzékenyítés, amellyel mesterséges fotoreceptorok hozhatók létre a retinában (39). A kutatócsoport leírta az ON-típusú bipoláris sejtek vagy retinalis ganglionsejtek csatornarodopszin általi depolarizációját, amely révén a retina felfogja a fényt és jellé alakítja (37, 38, 39, 40). Egy másik modellben a kutatócsoport a csap fotoreceptorokat célozta csendesítő optogenetikai reagens alkalmazása révén, így a csapok hiperpolarizálódtak és újra érzékennyé váltak a fényre. Ezek az eredmények az elsorvadt fotoreceptorral rendelkező fényérzés nélküli, retinitis pigmentosában vagy más öröklődő retinadisztrófiában szenvedő betegek kezelésében játszhatnak szerepet, akiknél a retinalis ganglionsejtek intaktak és vizuális információt tudnak továbbítani az agy felé (37, 38, 39, 40, 41, 42). Szintén tanulmányozták a pálcikák szerepét fotópikus körülmények között, és megállapították, hogy a pálcikák gátlóreléként működnek a csapokkal együttműködő horizontális sejtekre hatva (43). Roska Botond, Hillier Dániel és svájci kutatócsoportjuk aktívan együttműködnek Rózsa Balázs kutatócsoportjával, ami a Magyar Tudományos Akadémia Kísérleti Orvostudományi Kutatóintézetében közvetlenül vizsgálja kísérleti állatok látókérgét, és a szem által érzékelt vizuális információ megjelenését figyelik a neurális hálózatok aktivitásmintázataiban. Rózsa Balázs és kollégái fejlesztettek egy lézerrendszerrel összekötött szuperrezolúciós mikroszkópot, amely kihasználja a háromdimenziós leképezés előnyeit, több nagyságrenddel növeli az egyszerre vizsgálható sejtek számát és a vizsgálható térfogatot, továbbá a mérési sebességet. Segítségével lehetővé válik, hogy akár a videofelvételek képsebességével mérjék és módosítsák a sejtek működését. A kutatás folyamatában áttörést hozott, hogy Roska Botond másik hazai kollaborációs partnere, a Semmelweis Egyetem Anatómiai Szövet- és Fejlődéstani Intézetében dolgozó Szabó Arnold és kutatócsoportja kidolgozta a post mortem humán retinák hosszú távú (2-4 hét) életben tartásának módszerét, ezáltal lehetővé téve génmódosított sejtek részletes funkcionális vizsgálatát (44, 45, 46, 47).
A sikeres állatkísérleteket követően Roska Botond kutatócsoportja kollaborációban Jose-Alain Sahel és Serge Picaud kutatócsoportjával ex vivo humánretinákon is bizonyította, hogy a fotoreceptorok fényérzékennyé tehetők (39, 41). Az I. fázis klinikai tanulmány 2018-ban kezdődött (NCT03326336, PIONEER 2018-2020-2025) (48, 49), amelyben rekombináns adeno-asszociált vírusvektorral bevitt csatornarodopszin révén fényérzékennyé teszik a célsejteket, majd egy speciális stimuláló szemüveggel (Visual Interface Stimulating Glasses) felerősítik a külső vizuális jeleket az optogenetikailag módosított retina számára (39, 49). 2021 tavaszán nagy visszhangot keltett a publikáció, amelyben Usher-szindróma II. típusában szenvedő beteg látását sikerült részlegesen helyreállítani ezzel az optogenetikai módszerrel (49).
Jelenleg folyó másik optogenetikai módszert alkalmazó klinikai tanulmány 2015-ben kezdődött, várható befejezése 2035-ben lesz és szintén a retinitis pigmentosa kezelését célozza: RST-001 Phase I/II Trial for Advanced Retinitis Pigmentosa NCT02556736 (2015-2020-2035) (48, 50).
Szemészeti betegségeken kívül többek közt optogenetikai módszereket használtak fel gerincvelősérült egerek idegi funkcióinak felélesztésére, és a jutalommal, a motivációval és a félelemmel kapcsolatos jelutak szabályozására is. A kutatási eredmények biztatóak a Parkinson- és az Alzheimer-kór, az epilepszia, a skizofrénia, az ADHD, a különböző szenvedélybetegségek és a fájdalommal kapcsolatos rendellenességek területén is (50, 51, 52).
Genetikai alapú kutatások különböző betegségekben
Szemészeti vonatkozású kutatások CRISPR/Cas rendszerrel
Glaukóma
Egérmodellben sikerült megakadályozni a CRISPR/Cas9 rendszerrel a glaukóma kialakulását a myocilin gén inaktiválása révén (28). Egy másik egérmodellben a sugártest csarnokvíz-termelését és a szemnyomást csökkentették sikeresen intravitreális injekció révén bejuttatott CRISPR/Cas9 rendszer által az Aquaporin 1gén diszrupciójával (53, 54).
Öröklődő retinadisztrófiák
Leber congenitalis amaurosis CEP290 gén biallélikus mutációja okozta formájában (LCA10) a 26. intron mélyén elhelyezkedő mutációt (p.Cys998X, IVS26 c.2991+1655A>G), amely aberráns splice donor helyet hozott létre, sikerült szomatikus emlőssejteken és egérmodellben a CRISPR/Cas9 rendszer alkalmazásával kiiktatni, így normál CEP290 génexpressziót figyeltek meg (55). Az eredményes állatkísérletek után megkezdődött az oregoni Casey Eye Institute-ban az I/II. fázis klinikai tanulmány (BRILLIANCE study, NCT03872479, 2019-2024.), amelybe 18 LCA10 típusú, a specifikus intronikus mutációt hordozó beteg bevonását tervezik. A kezelés a fotoreceptorokat célozza subretinalis injekció alkalmazásával (48, 56, 57).
Az észak-amerikai Foundation Fighting Blindness jelenleg az alábbi CRISPR/Cas9 laboratóriumi kutatásokat támogatja: KCNJ13 gén okozta LCA, RP1-gén okozta pálcika-csap disztrófia, USH2A okozta retinadisztrófia, MYO7A-gén okozta Usher1B-szindróma (56).
Nedves típusú időskori makuladegeneráció
Egérmodellben alkalmazva a CRISPR/Cas9 rendszert sikerrel hoztak létre kis inszerciókat és deléciókat a Vegfa és Hif1-gének célhelyein, inaktiválva ezzel a Vegfa és Hif1-géneket. A tanulmány új génterápiás lehetőség hatásosságát írta le egereken, mivel nem alakult ki choroideális érújdonképződés (28).
Szemészeti vonatkozású kutatások és klinikai tanulmányok génterápiás vektorok alkalmazásával
Jelenleg (2023 októberében) egyetlen törzskönyvezett szemészeti génterápiás kezelés létezik: az RPE65-gén biallélikus mutációi okozta LCA/RP-kezelésére kifejlesztett Luxturna®. A fent leírt módszerekkel azonban számos kutatás, fejlesztés zajlik különböző betegségekben. Ezek nagyobb része még laboratóriumi, vagy állatkísérletes stádiumban van, de egyre nő a humán klinikai vizsgálati fázisba jutó fejlesztések száma is.
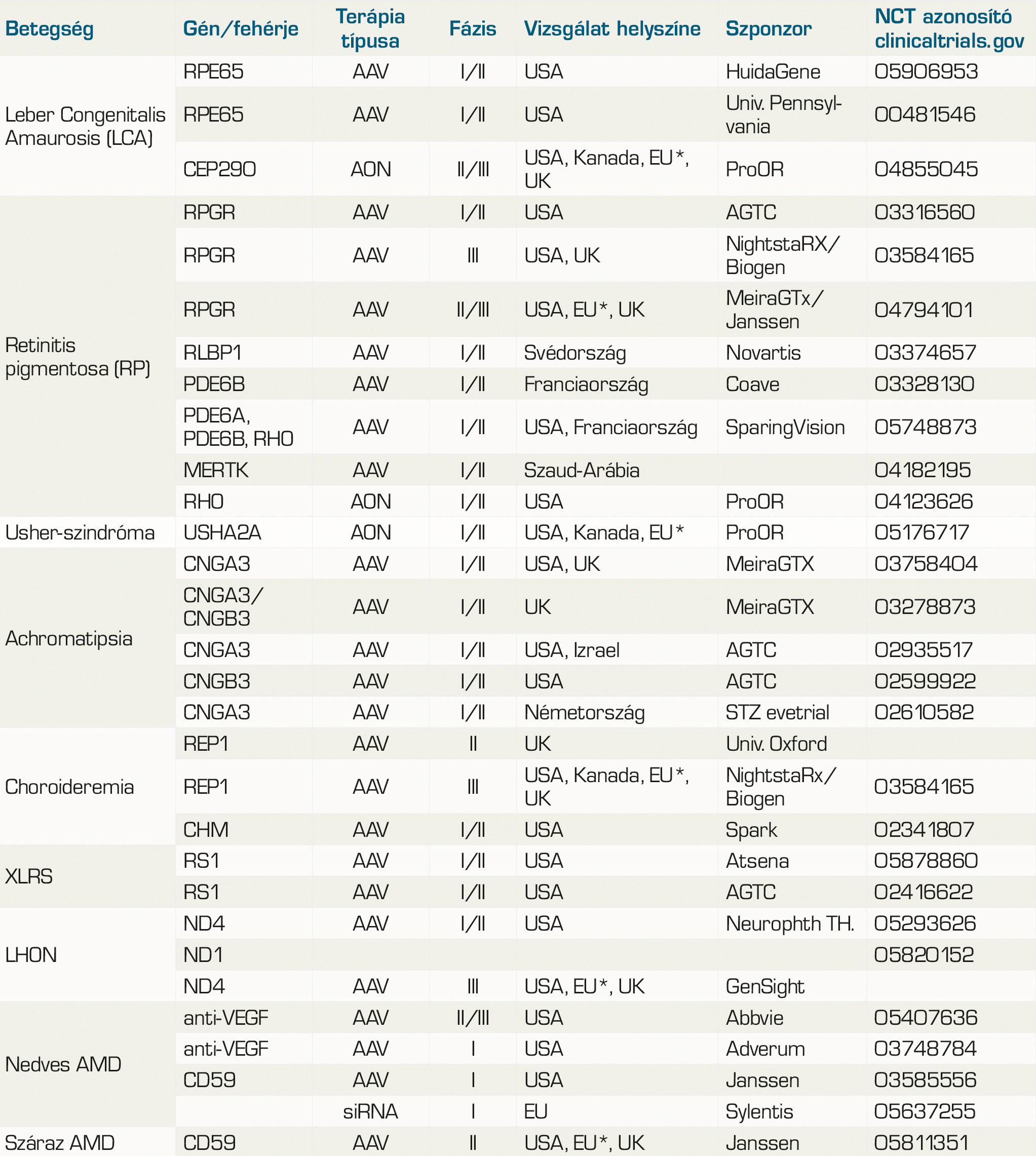
A klinikai vizsgálatokat nyilvántartó clinicaltrials.gov weboldalon számos paraméter szerint kereshetünk a jelenleg folyó vagy korábbi vizsgálatokra (48). 2019 áprilisában Trapani és Auricchio közleménye alapján 9 indikációban (achromatopsia, choroideremia, LCA, LHON, időskori makuladegeneráció, retinitis pigmentosa, Stargardt-betegég, Usher-szindróma, XLRS) összesen 63, jellemzően I-II. fázisú vizsgálat folyt örökletes retinadisztrófiák kezelése kapcsán (14). Egy évvel később, 2020-ban a Mária utcai füzetek Szemészeti Genetika számának publikálásakor már 70 találatot mutatott ugyanazon keresőszavakra a weboldal (58). Az indikációk lényegében nem változtak. Az azóta eltelt 3 évben sok vizsgálat befejeződött, új gyógyszeres kezelés azonban egyelőre nem kapott törzskönyvet.
A 2. táblázatban összefoglaltuk a jelenlegi, legfontosabbnak tartott szemészeti, genetikai alapú kezelésekkel kapcsolatos vizsgálatokat.
Leber Herediter Opticus Neuropathia kezelése kapcsán zajló kutatások
Sokáig úgy tűnt, hogy a második törzskönyvet kapó szemészeti génterápiás készítmény is egy Theodore Leber által leírt állapot gyógyítására alkalmas szer lesz. A LHON (Leber féle herediter opticus neuropathia, vagy örökletes szemidegbántalom) egy mitokondriális DNS-ben található patogén variáns (mutáció) okozta, maternális öröklődésmenetet mutató elváltozás (60).
A leggyakoribb LHON-variánst (G11778A) magába foglaló mitokondriális DNS-szakasz (ND4 komplex) génpótló kezelésére kifejlesztett lenadogen-nolparvovec (LUMEVOQ®, GS010; GenSight Biologics) törzskönyvezési vizsgálata lezajlott, az előzetes eredmények biztatóak voltak, de a hatásosság pontosabb megítélésre az Európai Gyógyszerügynökség (EMA) újabb, kiegészítő vizsgálat lefolytatását kezdeményezte.
Időskori makuladegeneráció (AMD)
Az öröklődő szembetegségek mellett a közeljövőben a klasszikusan szerzett (bár jelentős genetikai háttérrel rendelkező) betegségek kezelésében is szerepet kaphatnak a génterápiás eljárások. Jelenleg az időskori makuladegeneráció (AMD) nedves és száraz formájában is zajlanak humán klinikai vizsgálatok. A génterápiával módosított retinasejtekben termeltetett „endogen” anti-VEGF-molekulák egyszeri kezelés után hosszú távon biztosíthatják a megfelelő terápiát a nedves (neovaszkularizációs) AMD-ben szenvedő betegek számára, csökkentve ezzel az injekciókkal járó kockázatokat is (NCT05407636).
A száraz AMD késői stádiumával kapcsolatban (geografikus atrófia, GA) is léteznek kísérleti szerek: a Janssen cég intravitreálisan alkalmazott AAV vektoros készítményétől egyrészt az atrófiás területének növekedési ütemének, másrészt a nedves formába történő átmenet gyakoriságának a csökkenését várják (NCT05811351).
Látható, hogy számos génterápiás kutatás, fejlesztés zajlik, azonban a korábbi, 5-10 évvel ezelőtti helyzethez képest jelenleg lassulás figyelhető meg a biotechnológiai fejlesztések terén. A várva várt újabb gyógyszerek törzskönyvezése, forgalomba hozása megtorpant, egyelőre várnunk kell a következő génterápiákra. Ennek fő oka, hogy a mostanában befejezett humán génterápiás klinikai vizsgálatok eredményei elmaradnak az állatkísérletes modellek eredményei alapján elvárttól. Feltehető, hogy a vektorok és promoterek (szabályozó egységek) kifejeződésének hatékonysága, a génexpressziós különbségek, gén-gén interakciók állhatnak a háttérben. Ezek feltérképezéséhez és a tervezett terápia hatékonyságának növeléséhez jelenthet segítséget a posztmortem humán retina in vitro tenyésztése, a retina organoid modellek létrehozása, amely módszerek kifejlesztése Szabó Arnold kutatólaborjához fűződik.
Klinikai tanulmányok tervezésének egyik sarkalatos pontja a végpontok, vagyis az eredményességet bizonyító paraméterek meghatározása. Szemészetben gyakran a látóélességet (BCVA), vagy az OCT-vel mért retinavastagságot alkalmazzuk elsődleges végpontként. Öröklődő retinadisztrófiákban azonban a beteg gyakran olyan rossz látóélességgel bír, hogy a klasszikus vizsgálómódszerek (ETDRS-tábla) nem megfelelőek. Az ilyen klinikai vizsgálatokban speciális látóélesség-teszteket (low-luminance visual acuity, LLVA), objektív funkcionális módszereket (ERG, FST), vagy komplex mozgási feladatsort (multi-luminance Mobility Test, MLMT) szoktak végpontként választani (59).
Fontos, hogy bármely terápia során reális legyen a kitűzött cél. A túlzott – és be nem teljesülő – elvárások a betegek oldaláról csalódottsághoz, a kezelő orvosoknál frusztrációhoz, klinikai vizsgálat esetén pedig akár a törzskönyvezési eljárás elutasításához vezethetnek. A genetikai vizsgálatok előtérbe kerülése mellett újból egyre nagyobb hangsúlyt kap az alapos szemészeti kivizsgálás (deep phenotyping), mivel az adott beteg részletes anatómiai és funkcionális vizsgálata biztosít elég információt a terápiás indikációhoz és a várható javulás mértékének megállapításához.
Ahogy láthattuk, az új génterápiás eljárások kutatása egyre elterjedtebb, a biotechnológia ugrásszerű fejlődése azonban rengeteg etikai dilemmát is felvet. A génszerkesztő beavatkozások hosszú távú következményei még nem pontosan ismertek. Szomatikus sejtekben való alkalmazásuk más, mint ivarsejtekben. Az embriók génmódosításának engedélyezése még a téma szakértőit is megosztja, hiszen észrevétlen mutációk is előfordulhatnak az implantált génmódosított embriókban, ami komoly következményekkel járhat. Nem tudhatjuk, mi valósul meg 10-15 év múlva a fenti biotechnológiai lehetőségekből, és milyen hatással lesznek a jövőnkre.
Génterápiás kezelés hazánkban
RPE65-gén okozta Leber Congenitalis Amaurosis és Retinitis Pigmentosa kezelése
A szem immunprivilegizáltsága révén rendkívül jó génterápiás célszerv. Ezek egyik eredményeként kapott törzskönyvet a voretigene- neparvovec, amely az RPE65-gén biallélikus mutációi okozta Leber congenitalis amaurosis (LCA) és retinitis pigmentosa (RP, pálcika-csap disztrófia) kezelésére lett kifejlesztve. Az adeno-asszociált vírusvektorba csomagolt RPE65-gént tartalmazó gyógyszert a retinalis pigmenthámba kell bejuttatni altatásban végzett 23 G pars plana vitrectomia során subretinalis injekció formájában.
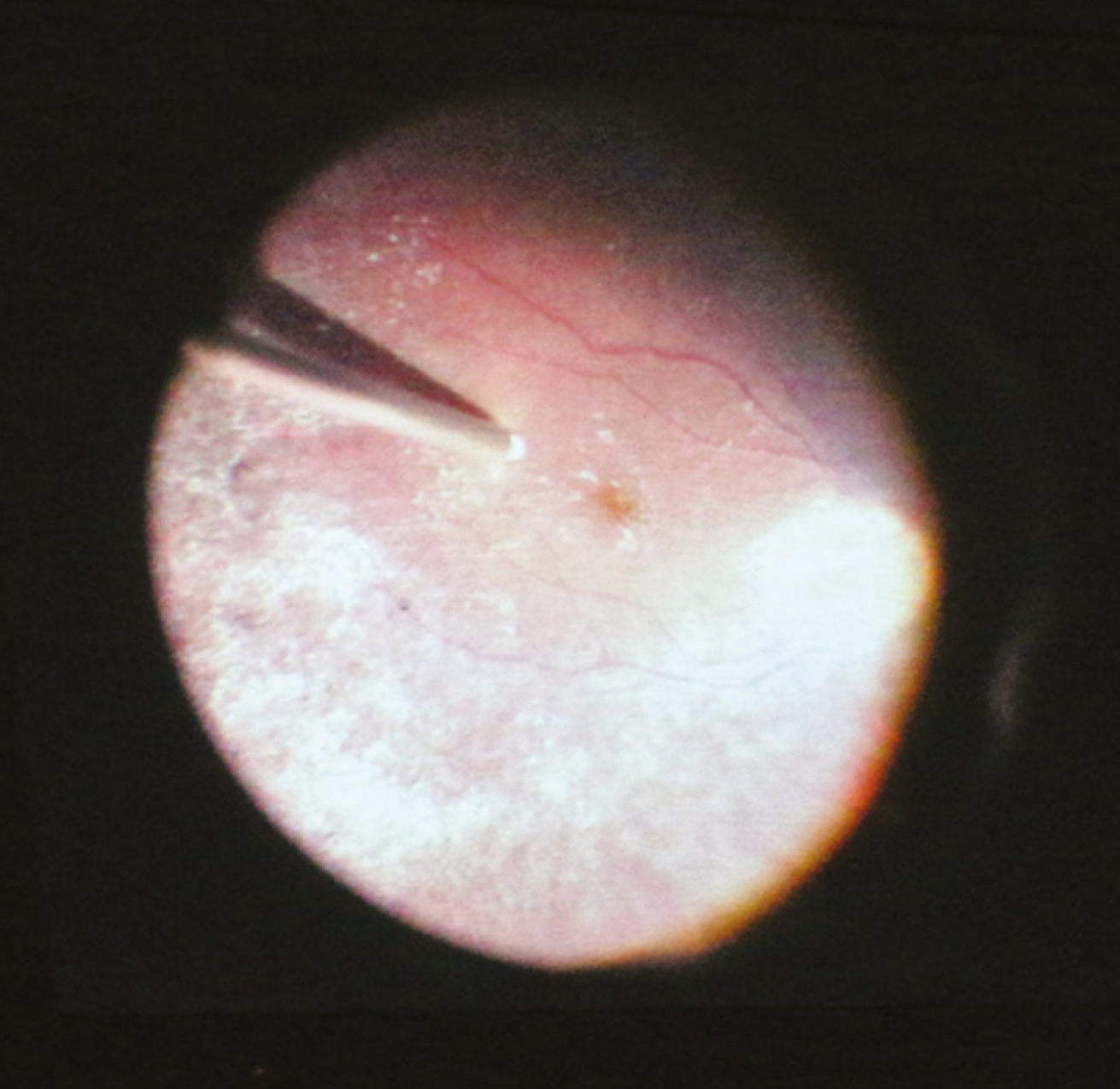
2022 júliusában a Semmelweis Egyetem Szemészeti Klinikája akkreditációt kapott, mint Szemészeti Génterápiás Centrum és az első év során megtörtént 10 kezelés LCA-ban szenvedő betegekben. A centrumban négy vitreoretinalis sebész, dr. Papp András, dr. Szabó Antal, dr. Barcsay György és dr. Resch Miklós részesült speciális képzésben. A gyógyszer a Semmelweis Egyetem Központi Gyógyszertárba egyéni megrendelés alapján a műtét előtti napon érkezik be egyedi NEAK-engedélyeztetést követően, a szállítás –80° történik a hűtési lánc szigorú ellenőrzésével. A Semmelweis Egyetem Gyógyszerészeti Intézetben történik a gyógyszer felolvasztása, illetve hígítása 2 órával a műtét előtt aszeptikus technikával és steril körülmények között a biztonsági előírásoknak megfelelő lamináris áramlású boxban. A képzett 6 gyógyszerész kolléga közül 2-2 fő vesz részt az aktuális subretinalis injekció elkészítésében, amely szobahőn tárolva a hígítástól számított 4 órán belül használható fel. A műtét során 1-4 retinotomiás nyílás készíthető a megfelelő dózis (300 ml) infúziós pumpával való bejuttatásához, a subretinalis injekció után folyadék-levegő csere történik. Az első 24 órában háton fekvő pozíciót javasolt a beteg számára. A perioperatív per os szteroidkezelést a műtét előtt 3 nappal szükséges megkezdeni 1 mg/ttskg/nap dózisban, de max. 40 mg/összdózisban adandó összesen 7 napig, majd 10 nap alatt felezve csökkentendő a megfelelő káliumpótlás és gyomorsavcsökkentő adása mellett (2, 3., 4. ábra).
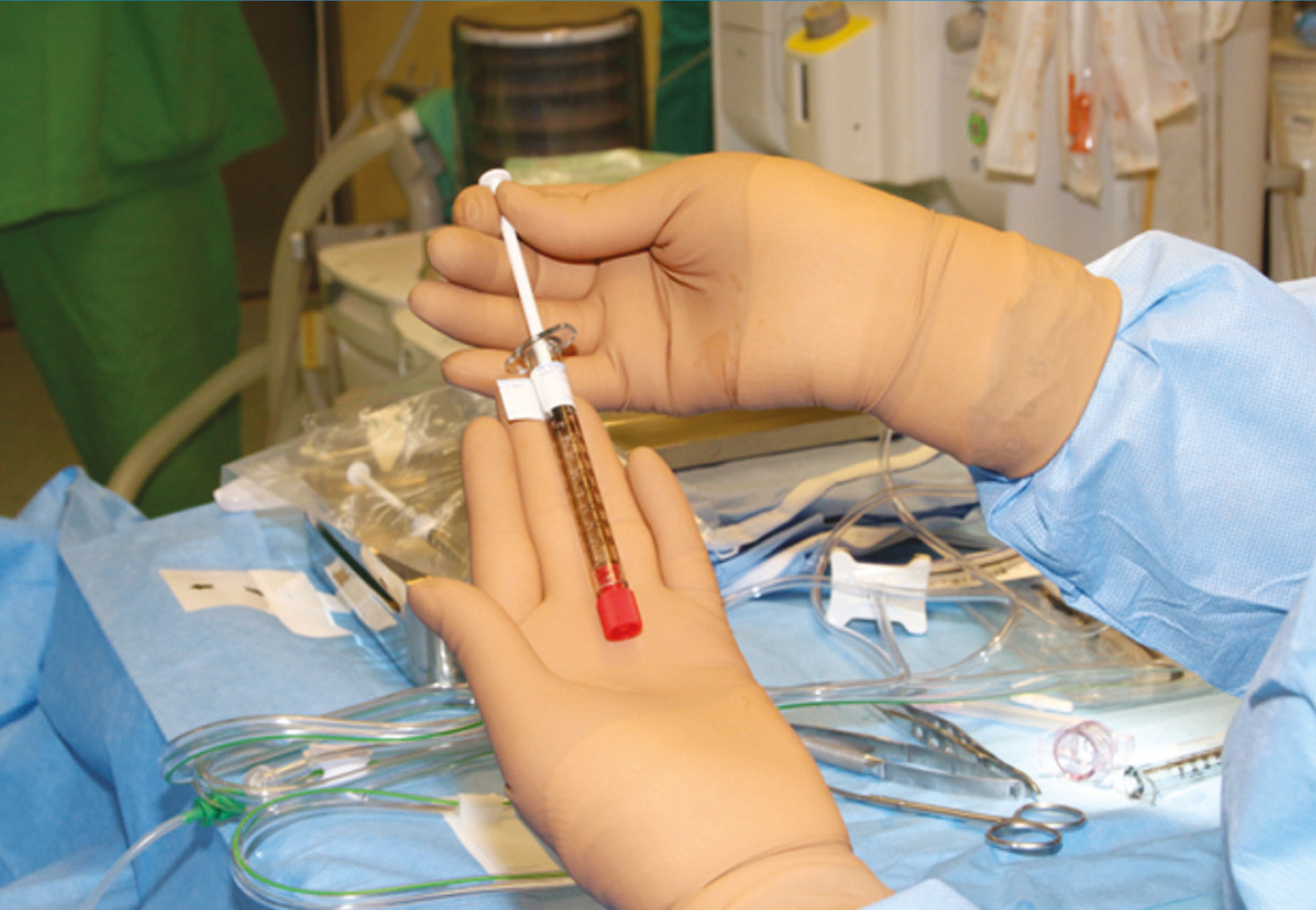
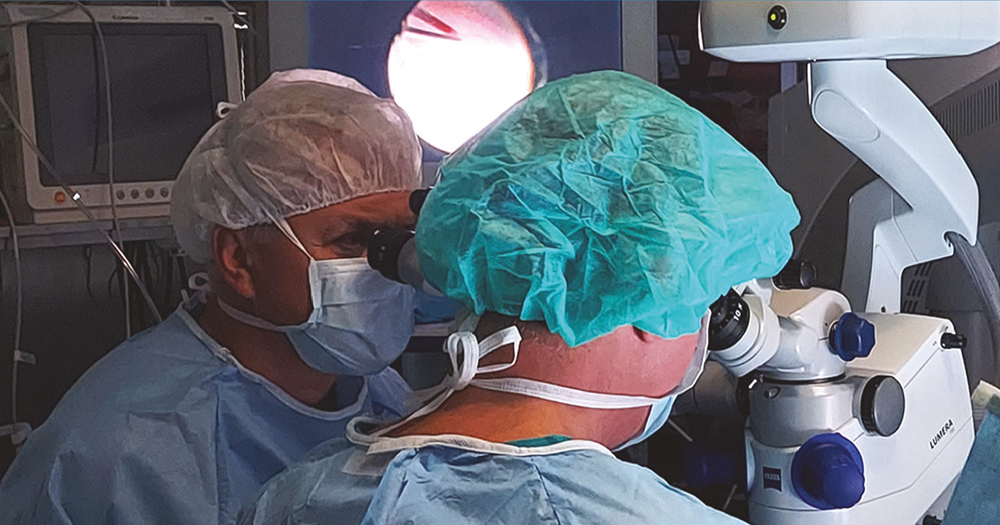
Összefoglalás
Az öröklődő retinadisztrófiák klinikailag és genetikailag rendkívül heterogén betegségcsoport. A leggyakoribb típusa a retinitis pigmentosa (pálcika-csap disztrófia), amelynek kóroki génjeit tekintve Európában magas a hordozók aránya, tehát nem ritkán előforduló kórkép.
A szem immunprivilegizáltsága miatt jó génterápiás target, emiatt számos kutatócsoport dolgozik lehetséges génterápia fejlesztésén az öröklődő retinadisztrófiák gyógyítására.
Ma már létezik törzskönyvezett gyógyszer (voretigene-neparvovec), amely az RPE65-gén biallélikus mutációi okozta Leber congenitalis amaurosis (LCA) és retinitis pigmentosa (RP, pálcika-csap disztrófia) kezelésére alkalmazható. Az adeno-asszociált vírusvektorba csomagolt RPE65-gént tartalmazó gyógyszert a retinalis pigmenthámba kell bejuttatni altatásban végzett pars plana vitrectomia során subretinalis injekció formájában. A gyógyszer hazánkban is elérhető, a 2022 nyarán megnyílt Szemészeti Génterápiás Centrumban, a Semmelweis Egyetem Szemészeti Klinikáján. A betegek genotipizálása és alapos fenotipizálása alapján betegregisztert hozunk létre, így a jövőbeli terápiás lehetőségek megjelenésekor ismert lesz az adott hazai betegcsoport, s ez meggyorsíthatja a kezelések alkalmazhatóságát. A szemészeti öröklődő betegségben szenvedő betegeknél érdemes részletes kivizsgálást és genetikai tanácsadást követően genetikai vizsgálatot végezni.
Nyilatkozat
A szerzők kijelentik, hogy speciális továbbképző közleményük megírásával kapcsolatban nem áll fenn velük szemben pénzügyi vagy egyéb lényeges összeütközés, összeférhetetlenségi ok, amely befolyásolhatja a közleményben bemutatott eredményeket, az abból levont következtetéseket vagy azok értelmezését.
Irodalom
1. Varsányi B. Genetika a szemészetben. Szemészet 2014; 151(4): 151–166.
2. Collins F S, Fink L. The Human Genome Project. Alcohol Health and Research World 1995; 19(3): 190–195.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6875757/
3. Chial H. DNA sequencing technologies key to the Human Genome Project. Nature Education 2008; 1(1): 219.
https//doi.org/10.1007/s13353-011-0057-x
4. Valkenburg D, van Cauwenbergh C, Lorenz B, et al. Clinical Characterization of 66 Patients With Congenital Retinal Disease Due to the Deep-Intronic c.2991+1655A>G Mutation in CEP290. Invest. Ophthalmol Vis Sci 2018; 59(11): 4384–4391.
https//doi.org/10.1167/iovs.18-24817.
5. Nassisi M, Mohand-Saïd S, Andrieu C, et al. Prevalence of ABCA4 Deep-Intronic Variants and Related Phenotype in An Unsolved “One-Hit” Cohort with Stargardt Disease. International Journal of Molecular Sciences 2019; 20(20).
https//doi.org/10.3390/ijms20205053
6. Sangermano R, Garanto A, Khan M, et al. Deep-intronic ABCA4 variants explain missing heritability in Stargardt disease and allow correction of splice defects by antisense oligonucleotides. Genet Med 2019 Aug; 21(8): 1751–1760. https//doi.org/10.1038/s41436-018-0414-9.
7. MacArthur D G, Balasubramanian S, Frankish A, et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science 2012 Feb 17; 335(6070): 823–8.
https//doi.org/10.1126/science.1215040
8. Richards S, et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17(5): 405–424.
https//doi.org/10.1038/gim.2015.30
9. Ganesh A, Keep RB. Genetic testing in retinal dystrophies. Oman J Ophthalmol 2011; 4(3): 105–107.
https//doi.org/10.4103/0974-620X.91264
10. Nash BM, Wright DC, Grigg RJ, et al. Retinal dystrophies, genomic applications in diagnosis and prospects for therapy. Transl Pediatrics 2015; 4 (2): 139–163.
https//doi.org/10.3978/j.issn.2224-4336.2015.04.03
11. Kállai-Mikola Gy. Látássérülés és rehabilitáció. Elemi rehabilitáció felnőtt korú látássérült személyek számára. Szemészet (megjelenés alatt)
12. Matthijs G, Souche E, Alders M, et al. EuroGentest. European Society of Human Genetics. Guidelines for diagnostic next-generation sequencing. Eur J Hum Genet 2016 Jan; 24(1): 2–5.
https//doi.org/10.1038/ejhg.2015.226.
13. https://net.jogtar.hu/jogszabaly?docid=a0800021.tv
14. Trapani I, Auricchio A. Has retinal gene therapy come of age? From bench to bedside and back to bench. Hum Mol Genet 2019; 28(R1): R108–R118.
https//doi.org/10.1093/hmg/ddz130
15. Boye SE, Alexander JJ, Witherspoon CD, et al. Highly Efficient Delivery of Adeno-Associated Viral Vectors to the Primate Retina. Hum Gene Ther 2016; 27(8): 580–597. h
https//doi.org/10.1089/hum.2016.085
16. Sadelain M, Riviere I, Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nature Reviews Cancer 2003; 3: 35–45.
https//doi.org/10.1038/nrc971
17. Curran KJ, Pegram HJ, Brentjens RJ. Chimeric antigen receptors for T cell immunotherapy: current understanding and future directions. J Gene Med. 2012 Jun; 14(6): 405–15. PMID: 22262649; PMCID: PMC4697438.
https//doi.org/10.1002/jgm.2604
18. Sahel JA, Dalkara D. Gene therapy for retinal dystrophy. Nature Medicine 2019; 25(2): 198–199.
https//doi.org/10.1038/s41591-019-0346-1
19. Murray SF, Jazayeri A, Matthes MT, et al. Allele-Specific Inhibition of Rhodopsin With an Antisense Oligonucleotide Slows Photoreceptor Cell Degeneration. Invest Ophthalmol Vis Sci 2015 Oct; 56(11): 6362–75.
https//doi.org/10.1167/iovs.15-16400.
20. Maeder M, Stefanidakis M, Wilson CJ, et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat Med 2019; 25(2): 229–233.
https//doi.org/10.1038/s41591-018-0327-9.
21. Arbabi A, Liu A, Ameri H. Gene Therapy for Inherited Retinal Degeneration. J Ocul Pharmacol Ther 2019; 35(2): 79–97.
https//doi.org/10.1089/jop.2018.008
22. Maguire AM, Simonelli F, Pierce EA, et al. Safety and efficacy of gene transfer for Leber's congenital amaurosis. N Engl J Med 2008; 358: 2240–2248. https//doi.org/10.1056/NEJMoa0802315
23. Russell S, Bennett J, Wellman J A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet 2017; 390: 849–860.
https//doi.org/10.1016/S0140-6736(17)31868-8
24. https: //hu.wikipedia.org/wiki/CRISPR
25. Barrangou R, et al. CRISPR Provides Acquired Resistance Against Viruses in Prokaryotes. Science 2007; 315: 1709–1712.
https//doi.org/10.1126/science.1138140
26. Ishino Y, et al. Nucleotide Sequence of the IAP Gene, Responsible for Alkaline Phosphatase Isozyme Conversion in Escherichia coli, and Identification of the Gene Product. Journal of Bacteriology 1987; 169: 5429–5433.
https//doi.org/10.1128/jb.169.12.5429–5433.1987
27. Jinek M, et al. A Programmable Dual-RNA-guided DNA Endonuclease in adaptive Bacterial Immunity. Science 2012; 337: 816–821.
https//doi.org/10.1126/science.1225829
28. Cong L, et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013; 339: 819–823.
https//doi.org/10.1126/science.1231143
29. Zetsche B, et al. Cpf1 is a Single RNA-guided Endonuclease of a Class2 CRISPR-Cas System. Cell 2015; 163: 759–771.
https//doi.org/10.1016/j.cell.2015. 09.038
30. Boyden E S, Zhang F, Bamberg E, et al. Millisecond-timescale, genetically targeted optical control of neural activity. Nature Neuroscience 2005; 8(9): 1263–1268.
https//doi.org/10.1038/nn1525
31. Chow BY, Boyden ES. Optogenetics and translational medicine. Sci Transl Med 2013; 5: 177ps5
https//doi.org/10.1126/scitranslmed.3003101
32. Nagel G, et al. Channelrhodopsin-2, a Directly Light-gated Cation-selective Membrane Channel. Proceedings of the National Academy of Sciences of the USA 2003; 100: 13940–13945.
https//doi.org/10.1073 pnas.193619210
33. Nagel G, et al. Light Activation of Channelrhodopsin-2 in Excitable Cells of Caenorhabditis elegans Triggers Rapid Behavioral Responses. Current Biology 2005; 15: 2279–2284
https//doi.org/10.1016/j.cub.2005.11.032
34. Zemelman BV, et al. Photochemical Gating of Heterologous Ion Channels: Remote Control over Genetically Designated Populations of Neurons. Proceedings of the National Academy of Sciences of the USA 2003; 100: 1352–1357.
https//doi.org/10.1073/pnas.242738899
35. Masland RH. The neuronal organization of the retina. Neuron 2012; 76: 266–280.
https//doi.org/10.1016/j.neuron.2012.10.002
36. Bi A, Cui J, Ma YP, et al. Ectopic expression of a microbial-type rhodopsin restores visual responses in mice with photoreceptor degeneration. Neuron 2006; 50: 23–33.
https//doi.org/10.1016/j.neuron.2006.02.026
37. Lagali PS, Balya D, Awatramani GB, et al. Light-activated channels targeted to ON bipolar cells restore visual function in retinal degeneration. Nat Neurosci 2008; 11: 667–675.
https//doi.org/10.1038/nn.2117
38. Busskamp V, Duebel J, Balya D, et al. Genetic reactivation of cone photoreceptors restores visual responses in retinitis pigmentosa. Science 2010; 329: 413–417.
https//doi.org/10.1126/science.1190897
39. Roska B. EMBO Mol Med. 2019; 11: e10218.
https//doi.org/doi.org/10.15252/emmm.201810218
40. Busskamp V, Roska B. Optogenetic approaches to restoring visual function in retinitis pigmentosa. Curr Opin Neurobiol 2011 Dec; 21(6): 942–6.
https//doi.org/10.1016/j.conb.2011.06.001
41. Busskamp V, Picaud S, Sahel JA, Roska B. Optogenetic therapy for retinitis pigmentosa. Gene Therapy 2012; 19(2): 169–175.
https//doi.org/10.1038/gt.2011.155
42. Cronin T, Vandenbergher L H, Hantz P, et al. Efficient transduction and optogenetic stimulation of retinal bipolar cells by a synthetic adeno-associated virus capsid and promoter. EMBO Mol Med 6 2014; 1175–1190. https//doi.org/10.15252/emmm.201404077
43. Szikra T, Trenholm S, Drinnenberg A, et al. Rods in daylight act as relay cells for cone-driven horizontal cell-mediated surround inhibition. Nat Neurosci 2014; 17: 1728–1735.
https//doi.org/10.1038/nn.3852
44. Katona G, Szalay G, Maák P, et al. Fast two-photon in vivo imaging with three-dimensional random-access scanning in large tissue volumes. Nat Methods 2012; 9(2): 201–8.
https//doi.org/10.1038/nmeth.1851
45. Hillier D, Fiscella M, Drinnenberg A, et al. Causal evidence for retina-dependent and -independent visual motion computations in mouse cortex. Nat Neurosci 2017; 20(7): 960–968.
https//doi.org/0.1038/nn.4566
46. Wertz A, Trenholm S, Yonehara K, et al. PRESYNAPTIC NETWORKS. Single-cell-initiated monosynaptic tracing reveals layer-specific cortical network modules. Science 2015; 349(6243): 70–4.
https//doi.org/10.1126/science.aab1687
47. Jüttner J, Szabo A, Gross-Scherf B, et al. Targeting neuronal and glial cell types with synthetic promoter AAVs in mice, non-human primates and humans. Nat Neurosci 2019; 22(8): 1345–1356.
https//doi.org/10.1038/s41593-019-0431-2
48. www.clinicaltrials.gov
49. Sahel J A, Boulanger-Scemama E, Pagot C, et al. Partial recovery of visual function in a blind patient after optogenetic therapy. Nat Med 27 2021; 1223–1229.
https//doi.org/10.1038/s41591-021-01351-4
50. Simunovic MP, Shen W, Lin JY, et al. Optogenetic approaches to vision restoration. Experimental Eye Research 2018;
https//doi.org/10.1016/j.exer.2018.09.003
51. Chemi G, Brindisi M, Brogi S, et al. A light in the dark: state of the art and perspectives in optogenetics and optopharmacology for restoring vision. Future Med Chem 2019; 11(5): 463–487.
https//doi.org/10.4155/fmc-2018-0315
52. Farnum A, Pelled G. New Vision for Visual Prostheses. Front Neurosci 2020; 14: 36.
https//doi.org/10.3389/fnins.2020.00036
53. Jain A, Zode G, Kasetti R B, et al. CRISPR-Cas9–based treatment of myocilin associated glaucoma. PNAS 2017; 114(42)11199–11204.
https//doi.org/10.1073/pnas.1706193114
54. Wu J, Bell OH, Copland DA, et al. Gene Therapy for Glaucoma by Ciliary Body Aquaporin 1 Disruption Using CRISPR-Cas9. Molecular Therapy 2020; 28(3): 820–829.
https//doi.org/10.1016/j.ymthe.2019.12.012
55. Maeder M, Stefanidakis M, Wilson CJ, et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat Med 2019; 25(2): 229–233.
https//doi.org/10.1038/s41591-018-0327-9
56. www.fightingblindness.org
57. Zetsche B, Volz SE, Zhang F. A split-Cas9 architecture for inducible genome editing and transcription modulation. Nat Biotechnol 2015; 33(2): 139–42.
https//doi.org/10.1038/nbt.3149
58. Varsányi B, Vámos R, Lesch B, et al. Génterápiás klinikai tanulmányok az öröklődő szemészeti kórképekben. Mária Utcai Füzetek. A szemészeti genetika aktualitásai 2020; VI (3): 27–32. ISSN 2416-240X
59. Csaky K, Ferris 3rd F, Chew E Y, et al. Report From the NEI/FDA: Endpoints Workshop on Age-Related Macular Degeneration and Inherited Retinal Diseases. Invest Ophthalmol Vis Sci 2017; 58(9): 3456–3463.
https//doi.org/10.1167/iovs.17-22339
60. Knézy K, Vajda Sz, Keglevich A, et al. Leber-féle örökletes látóideg bántalom (LHON). Szemészet 2023; 160(1): I–VII.
61. De Angeli P, Reuter P, Hauser S, et al. Effective splicing restoration of a deep-intronic ABCA4 variant in cone photoreceptor precursor cells by CRISPR/SpCas9 approaches. Mol Ther Nucleic Acids 2022 Jul 31; 29: 511–524. https//doi.org/10.1016/j.omtn.2022.07.023