Bardet–Biedl syndrome – Case report of two patients diagnosed with the BBS10 gene variant
doi: 10.55342/szemhungarica.2023.160.3.122
Original scientific paper
Summary
Introduction: Bardet–Biedl syndrome (BBS) is a rare multisystemic autosomal recessive disease that belongs to the group of ciliopathies. It presents ophthalmologically as syndromic retinitis pigmentosa. The aim is to describe the characteristics of the BBS10 subtype by presenting two cases.
Case report: At the University of Debrecen, Department of Ophthalmology, at the end of 2022, sixty patients with previously diagnosed retinitis pigmentosa had a detailed ophthalmological examination and genetic sampling for genotyping (Blueprint Genetics, Retinal Dystrophy Panel Plus, version 7). Biallelic mutations in the BBS10 gene were found in two male patients, confirming Bardet–Biedl syndrome. Polydactyly, renal failure, retinal dystrophy, and obesity were present in both patients. In addition, learning difficulties and hypogonadism were observed in one of them.
Conclusion: Based on the literature and our clinical findings, the BBS10 subtype typically presents with severe Bardet-Biedl syndrome features such as visual disturbances, renal impairment, and obesity.
Összefoglaló
Bevezetés: A Bardet–Biedl-szindróma (BBS) egy ritka multiszisztémás autoszomális recesszíven öröklődő betegség, amely a ciliopátiák közé tartozik. Szemészeti szempontból szindrómás retinitis pigmentosa képében jelenik meg. A közlemény célja a BBS10-altípus jellemzőinek ismertetése két eset bemutatása kapcsán.Esetismertetés: A Debreceni Egyetem Szemklinikáján, 2022 végén hatvan, korábban degeneratio pigmentosa retinaevel diagnosztizált beteg részletes szemészeti vizsgálatát és genetikai mintavételét végeztük el, amelyek eredményeképpen a betegek genotipizálása megtörtént (Blueprint Genetics, Retinal Dystrophy Panel Plus, version 7). Két férfi beteg esetében a BBS10 gén biallélikus mutációja igazolódott és ezzel a Bardet–Biedl-szindróma megerősítést nyert. Polydactylia, veseelégtelenség, retinadisztrófia és elhízás mindkét betegnél jelen volt. Emellett az egyik betegnél tanulási nehézséget és hypogonadismust is tapasztaltunk.
Megbeszélés: Az irodalmi adatokat és a klinikumot összevetve megállapíthatjuk, hogy a BBS10-altípusnál jellemzően súlyos formában jelentkeznek a Bardet–Biedl-szindróma jellemzői, úgymint a látászavar, vesekárosodás és az elhízás.
Keywords
Bardet–Biedl syndrome, Degeneratio pigmentosa retinae, BBS10 gene
Kulcsszavak
Bardet–Biedl-szindróma, degeneratio pigmentosa retinae, BBS10 gén
Bevezetés
A Bardet–Biedl-szindróma (BBS) egy ritka, súlyos, több szervet érintő genetikai rendellenesség. Gyakorisága Európában és Észak-Amerikában 1:100 000 alá esik. Kelet-Európában eddig csak néhány esetről számoltak be, azonban a régióban még nem végeztek szisztematikus, genotipizálással egybekötött BBS-vizsgálatokat (7).
A BBS diagnosztikai kritériumait Beales és munkatársai a BBS-gének felfedezése előtt a klinikai kép alapján határozták meg (4). A BBS diagnózisát azoknál a betegeknél állíthatjuk fel, akik a betegség 6 elsődleges jellemzőjéből legalább 4-et, vagy 3 elsődleges jellemző mellett 2 másodlagos jellemzőt hordoznak. Elsődleges klinikai jellemzők közé tartozik a retinadisztrófia, az elhízás, a polydactylia, a vesefunkció-zavar, a különböző tanulási nehézségek és a hypogonadismus (1. ábra).
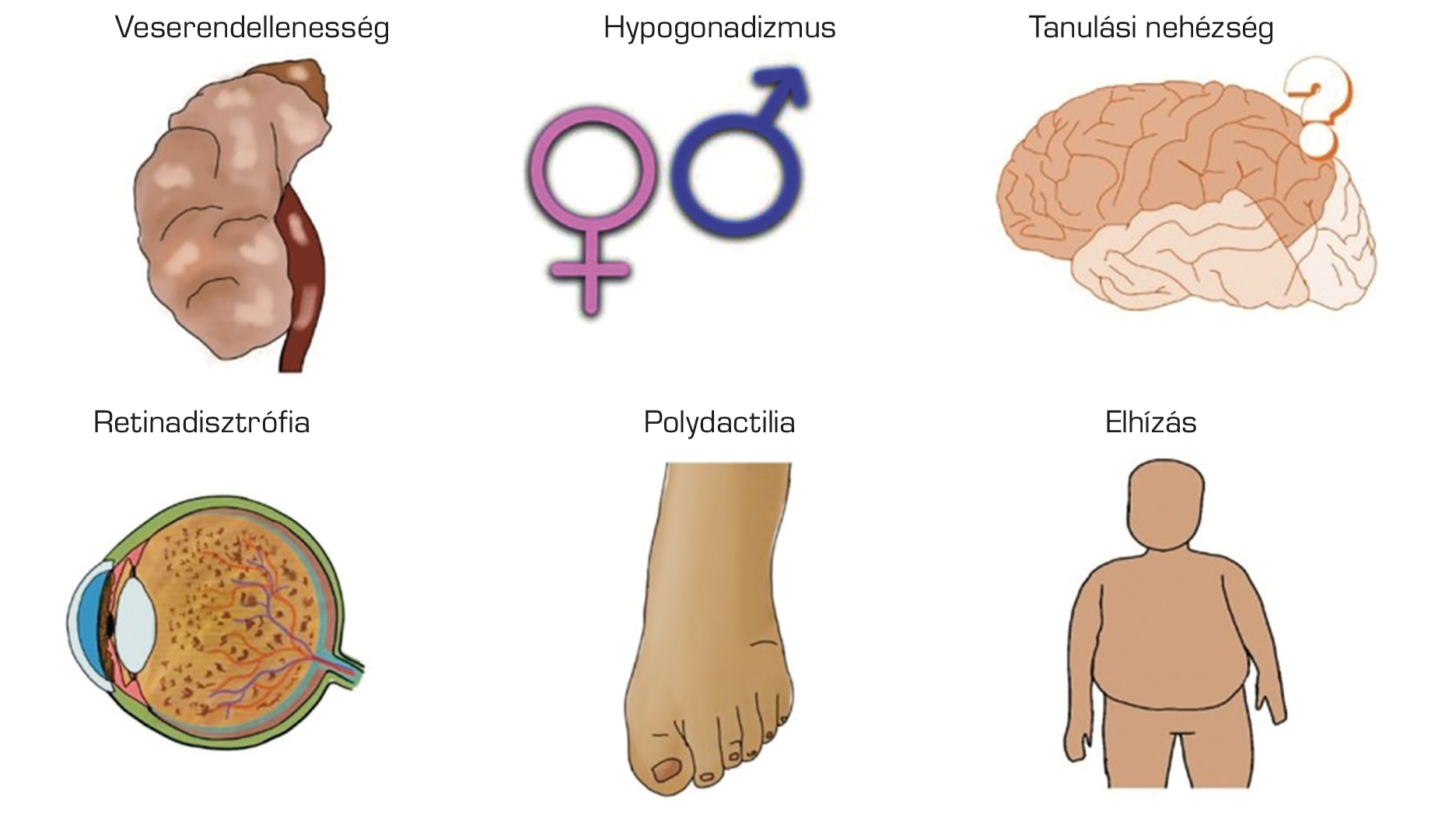
Másodlagos jellemzők a fejlődési zavar, a beszédzavar, a brachydactylia vagy syndactylia, a fogászati rendellenességek, az ataxia, a gyenge koordináció, a szaglási zavar, a diabetes mellitus és a veleszületett szívbetegség. Egyes szerzők a magas vérnyomást, a májrendellenességeket, a bronchialis asztmát, a középfülgyulladást, a náthát, a koponya-arc diszmorfizmust is a szindróma tünetének tartják (7). Szemészeti tünetek közül kiemelendő a fotoaverzió és a nyctalopia, amely általában 8 éves korban jelentkezik, és körülbelül 15–20 éves korban a látás elvesztéséig súlyosbodik (1). A BBS elsősorban pálcika-csap disztrófia, azonban előfordulhat, hogy elsődlegesen a csapok érintettek és ezt követik a pálcikák degenerációja. Gyakori szemrendellenességek a szürkehályog és fénytörési hibák is (4).
Biallélikus variánsokat 26 BBS-génben azonosítottak, amelyek a szindróma különböző típusait okozhatják (2). A BBS-gének olyan fehérjéket kódolnak, amelyek részt vesznek a csillók (ciliumok) biogenezisében és működésében (1). A BBS1, BBS2, BBS4, BBS5, BBS7, BBS8, BBS9 és BBS18-fehérjék a BBSome összetevői, amely az intraflagelláris transzportmolekulák számára adapterként működik. A BBS6, BBS10 és BBS12-fehérjék chaperonszerű komplexet alkotnak, amely fontos szerepet játszik a BBSome összeszerelésében (2). A BBS-gének közül a BBS1 és a BBS10 a leggyakrabban érintettek (6). A BBS10 gén mutációja a 10-es típusú Bardet–Biedl-szindrómát okozza. A becslések szerint az összes eset 21%-át teszik ki. A mutációk különböző típusait írták le, amelyekből a leggyakoribbak a missense (aminosavcserével járó) mutációk és a kis deléciók (2).
Esetismertetés
A Debreceni Egyetem Szemklinikáján 2022. szeptember és december között hatvan, korábban retinitis pigmentosával diagnosztizált beteg részletes szemészeti vizsgálatát és genetikai mintavételét végeztük el, amelyek eredményeképpen a betegek genotipizálása megtörtént (Blueprint Genetics, Retinal Dystrophy Panel Plus, version 7). Közülük két beteg esetében a genetikai vizsgálat bizonyította a már korábban felállított BBS jelenlétét. A szemészeti vizsgálat (látásélesség-meghatározás, biomikroszkópos vizsgálat oftalmoszkópiával, fundusfotó, OCT) mellett a betegek részletes anamnézisének áttekintése is megtörtént.
1. eset
A 40 éves férfi beteg családi anamnézise negatív volt, és az elsődleges klinikai képet a polydactylia, a retinitis pigmentosa, az elhízás és a vesekárosodás jellemezte. A BBS diagnózisa 20 éves korában került megállapításra. Csecsemőkorában polydactylia miatt mind a négy végtagjáról a 6. ujjat eltávolították (2. ábra).
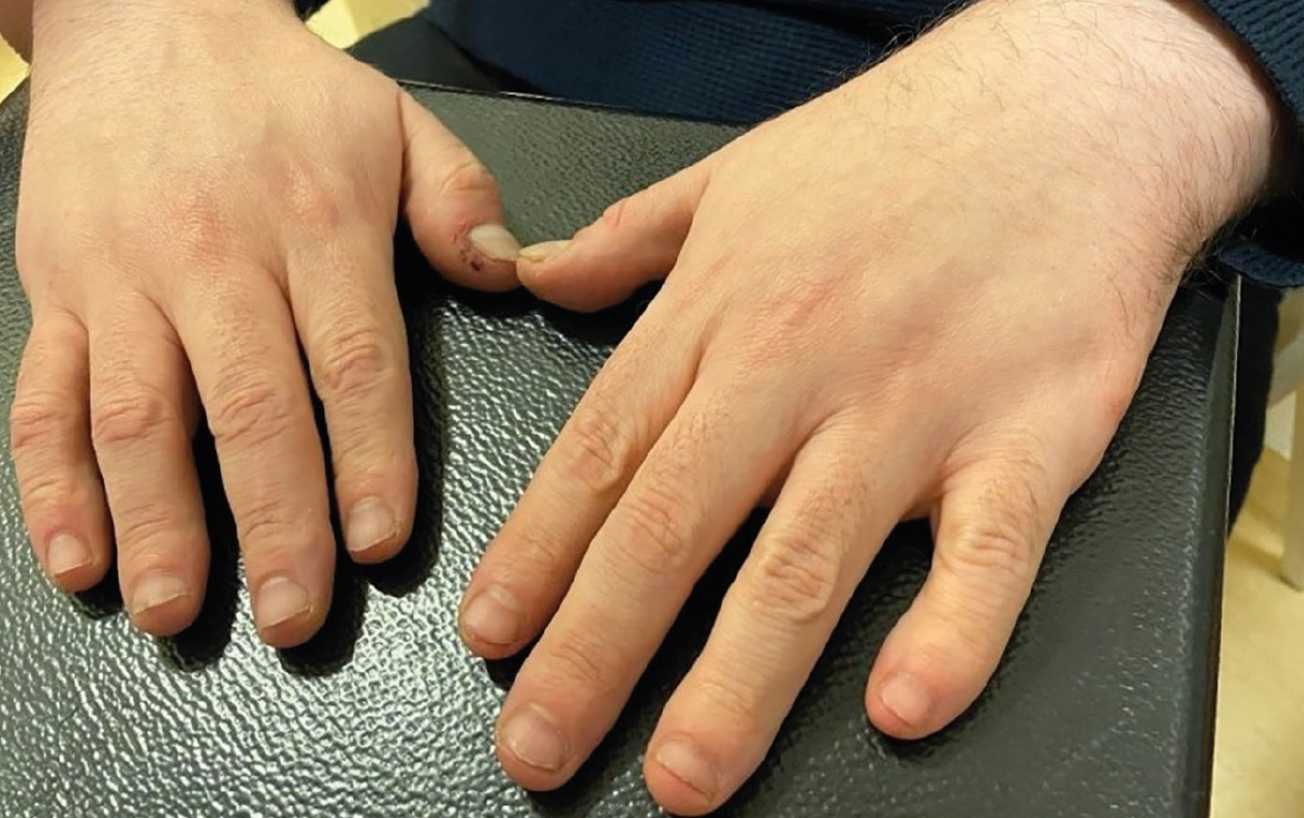
Vizsgálata során mindkét lábon a II-III. ujj syndactyliáját figyeltük meg. Obesitas miatt már 6 éves korától vizsgálták, eddigi legnagyobb súlya 130 kg (BMI: 46,6) volt. Szemklinikai vizsgálata során súlya 105 kg, magassága 167 cm, így a testtömegindexe (BMI) 37,7 volt. Súlyproblémáival összefüggésben nem inzulindependens diabétesz jelentkezett. Krónikus veseelégtelenség miatt 30 éves korától művesekezelésben részesült, majd 36 évesen vesetranszplantáció történt. Hyperparathyreosis miatt mind a négy mellékpajzsmirigy eltávolításra került, amelyet részleges autotranszplantáció követett a musculus sternocleidomastoideusba. Hipertónia miatt 27 éves korától részesül gyógyszeres kezelésben, emellett hyperurikaemia és hyperlipidaemia is ismert. Betegünk intellektusa kiváló, jogi diplomával rendelkező könyvtári informatikus. Megjelenését centrális elhízás, diszmorf arc és frontális kopaszodás jellemezte. Szemészeti anamnéziséből kiderült, hogy sötétben már gyermekkora óta rosszul lát. 17 évesen a legjobb korrigált látóélessége még 0,5/0,5 volt, majd 24 éves korára már mind a két szemén csupán fényérzést és kézmozgást tapasztalt.
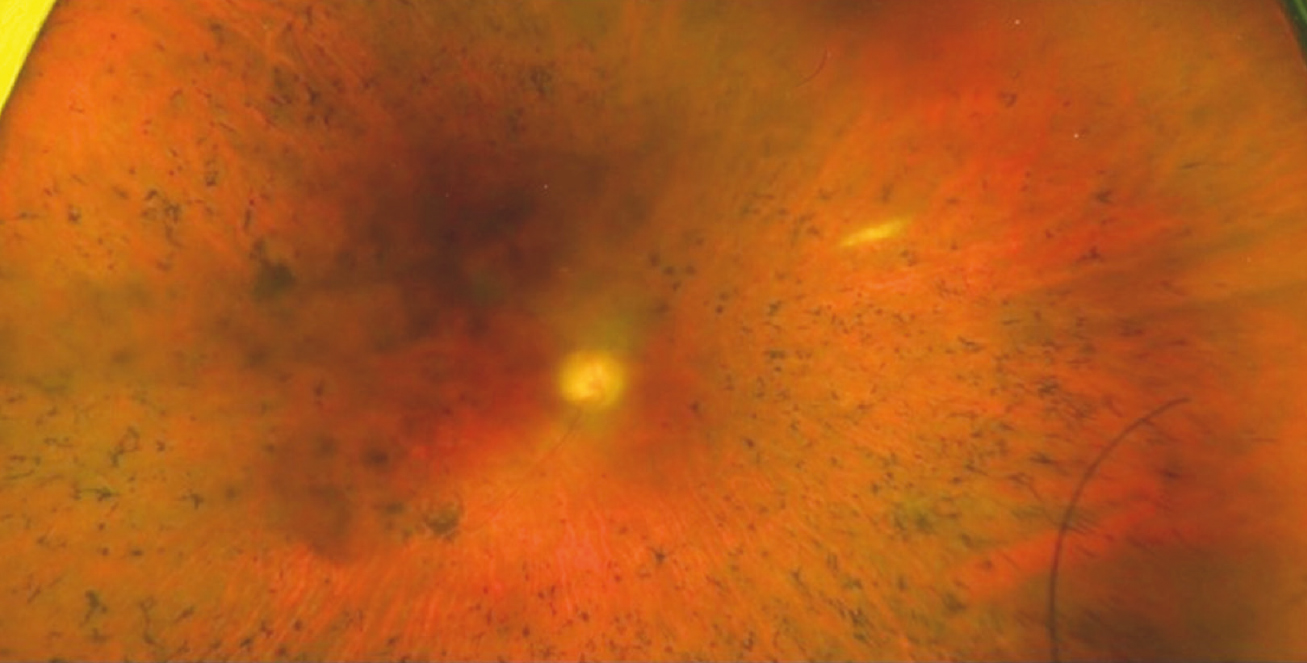
Vizsgálatunk során a beteg csupán kézmozgást érzékelt. Szemmozgások vizsgálata során kereső nystagmust tapasztaltunk. A szemlencse átvilágításakor hátsó, illetve elülső kérgi homályok voltak láthatók, üvegtesti töredezettség mellett. Szemnyomása 19-20 Hgmm. Oftalmoszkópos vizsgálat során tágításban kifejezetten halvány, atrófiás papillákat, illetve a retinális perifériát és a centrumot is érintő típusos csontsejtszerű pigmentdegenerációt láthattunk (3. ábra).
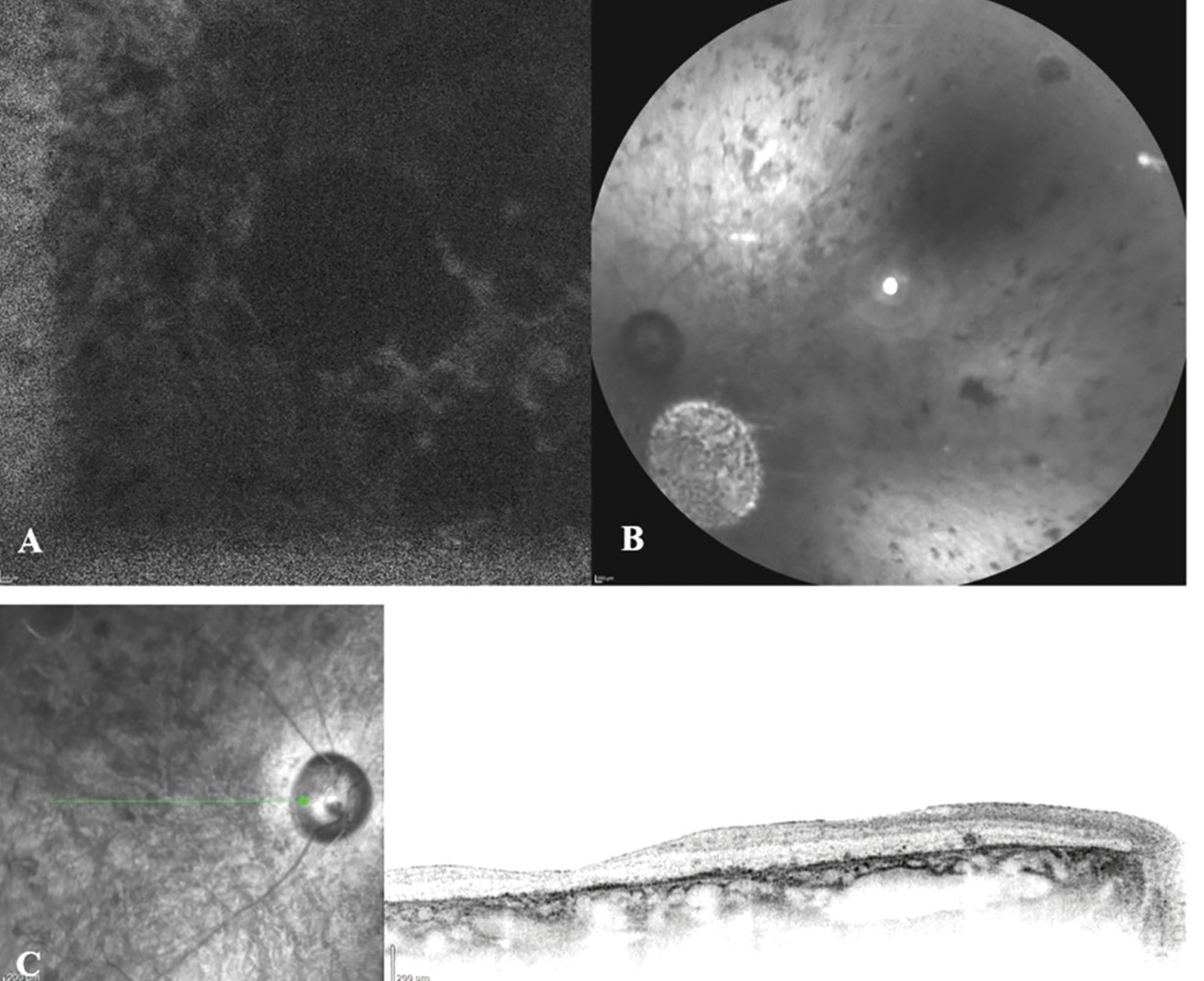
Heidelberg Spectralis OCT-vel a neuretina szerkezetének felbomlását, kifejezett atrófiát és pigmentkicsapódásokat figyelhettünk meg. A fundus autofluoreszcencia kifejezetten csökkent volt (4. ábra).
A genetikai vizsgálat két heterozigóta missense variánst azonosított a BBS10 génben: c.258T>A, p.(Phe86Leu) és c.1804G>C, p. (Val602Leu), autoszomális recesszív öröklésmenettel. A két nukleotideltérés egyelőre bizonytalan jelentőségű variánsként (VUS = variant of uncertain significance) klasszifikálható, mivel nem szerepel a mutációs adatbázisokban, azonban a betegnél talált tünetegyüttes alátámasztja az új generációs szekvenálás eredményét.
2. eset
A 49 éves férfi betegünk kórelőzményében obesitas, degeneratio pigmentosa retinae (DPR), polydactylia, hypogonadismus, hyperlipoproteinaemia, hipertónia, empty sella és veseelégtelenség szerepeltek. A páciensünk a BBS hat elsődleges tünete közül mind a hattal rendelkezett, és a BBS diagnózis felállítása 10–15 éves kora között megtörtént. A betegtől felvett anamnézis alapján 2 éves korában izomgyengeség miatt dokumentációval alá nem támasztott konzervatív kezelés történt. A páciens vesegondjai diabetes insipidus formájában már csecsemőkorban elkezdődtek. A glomeruláris filtrációs ráta (GFR) 33 éves korában 21 ml/perc volt, majd 36 éves korától már művesekezelésre szorult. Vizsgálatakor 7 ml/perc GFR mellett a vesetranszplantációt negálta. Vesebetegség anyai ágon 2 unokatestvérnél enyhe formában ismert. Az alsó végtagi polydactylia (5. ábra) miatt speciális lábbelit viselt, azonban a 6. ujjak megjelenése szabályos, és az eltávolítását korábban sem ajánlották az esetlegesen talpon kialakuló egyensúlyzavar miatt.
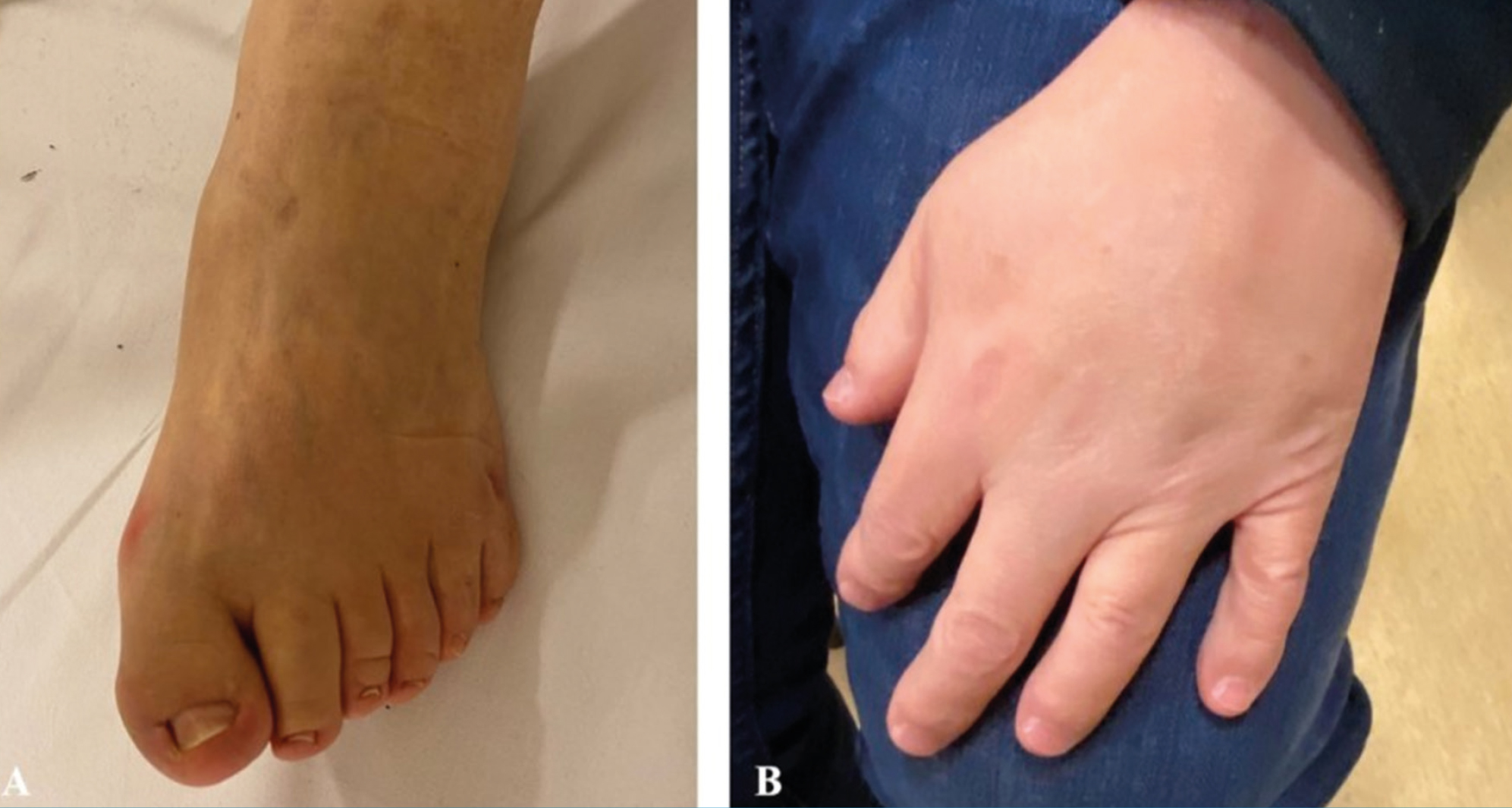
Szemészeti vizsgálatakor testsúlya 83,5 kg, a magassága 168 cm, és a BMI: 29,6 kg/m2 volt. Legnagyobb eddigi testsúlya 109 kg volt (BMI: 38,6). A szülők elmondása alapján a középiskola alatt tanulási nehézségei voltak.
Páciensünknél 4 éves korában a szemfenéken DPR-re utaló jeleket még nem írtak le, azonban 5 évesen már beszűkült látótér, megnyúlt látenciájú VEP és kioltott ERG dokumentált. 7 éves kora körül éjszaka már gondjai voltak a látással, a DPR diagnózisát kilenc éves korában 20°-ig koncentrikusan beszűkült látótér mellett felállították, strabismus divergens mellett. Az évek során szemészeti státusza progrediált. 20 éves kora körül jelentkezett nystagmusa, 23 évesen a legjobb látóélessége 1,0/0,3, majd 37 évesen már csak 0,2/0,3 volt.
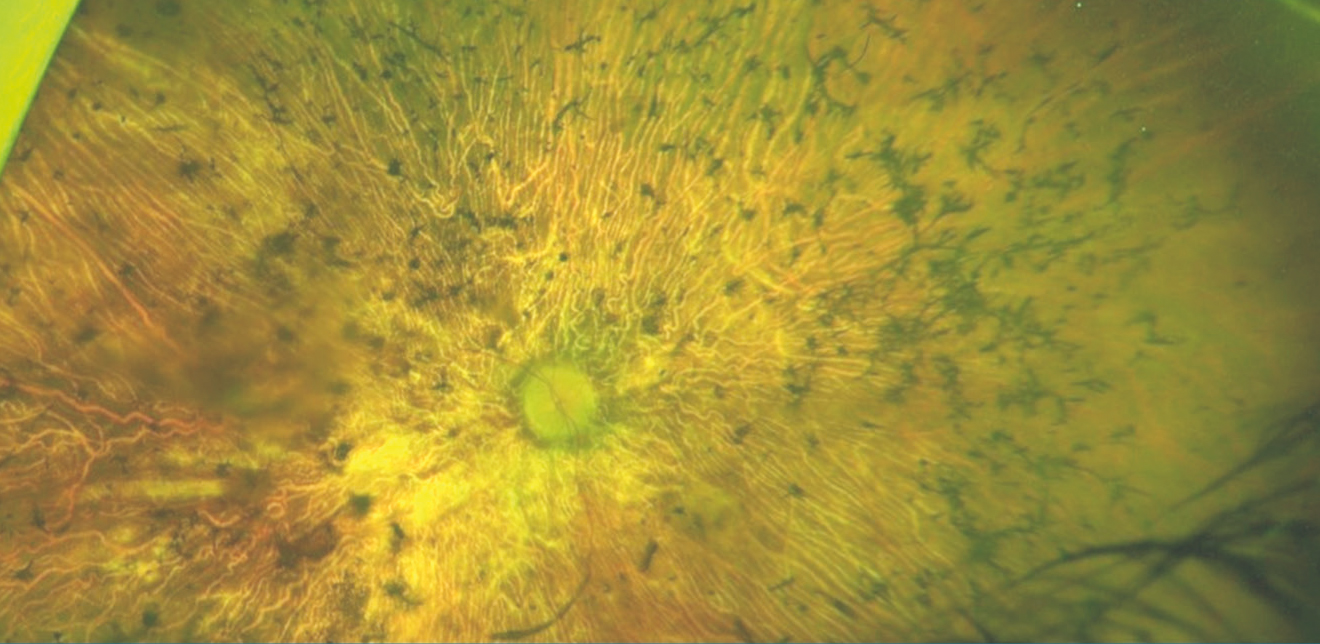
A genetikai mintavételkor csupán kézmozgás látással rendelkezett. Szemmozgások vizsgálatakor horizontális nystagmust észleltünk. A szemlencse átvilágításakor mindkét oldalon hátsó kérgi katarakta és töredezett üvegtest volt látható, kompenzált szemnyomás mellett. Oftalmoszkópos vizsgálat során a retina kifejezetten sorvadt volt, táblázott fundus, illetve a csontsejtszerű és paravasalis pigmentkicsapódások látszódtak. A DPR súlyos és típusos, érintett a perifériás retina és makula is (6. ábra).
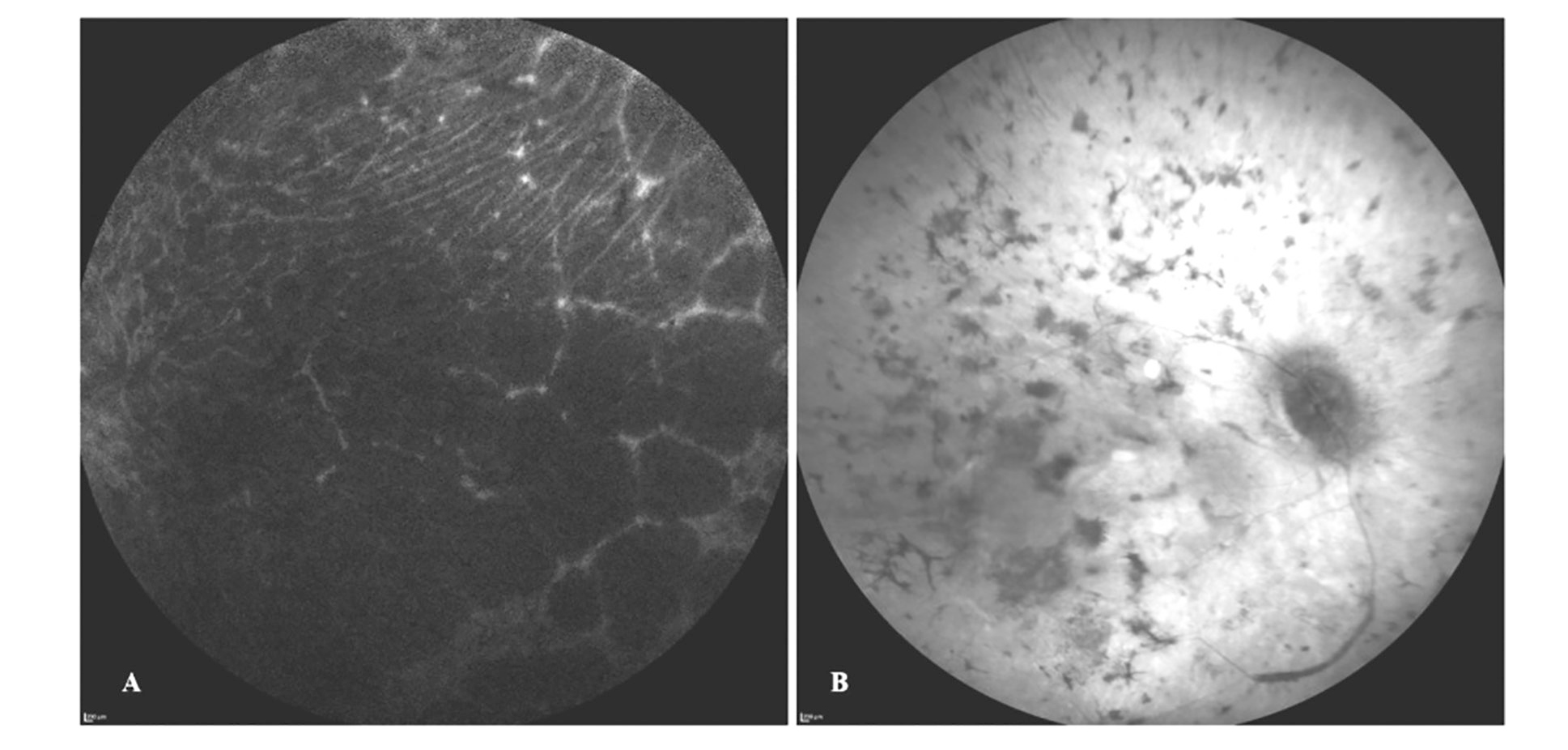
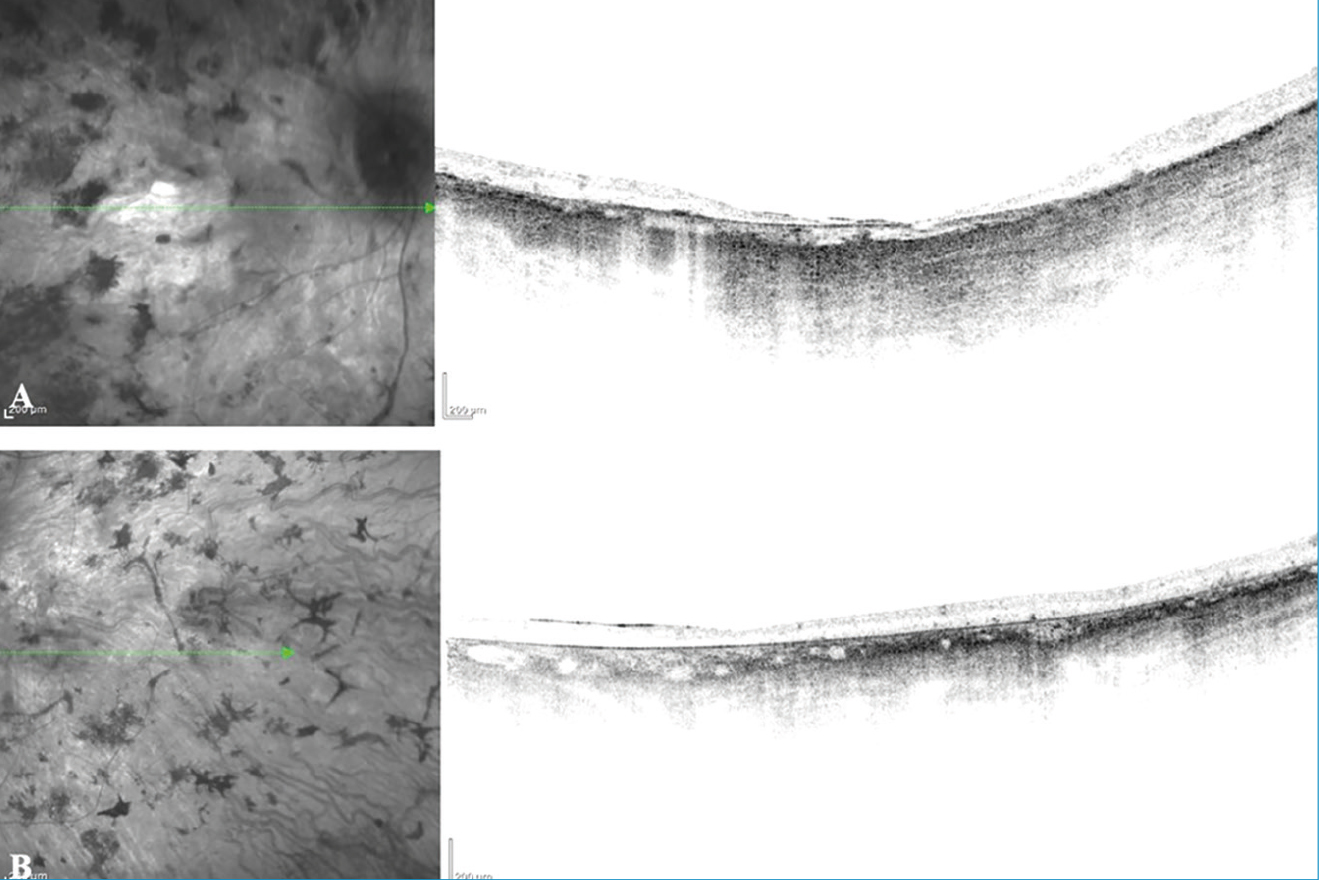
Heidelberg Spectralis OCT-vel való vizsgálatakor fixációs nehezítettséget tapasztaltunk a gyenge vízus és a nystagmus miatt. Jobb szemén a makulatájon kifejezett elvékonyodást láthattunk, mindkét szemen a neuroretina rétegeinek kifejezett károsodását, illetve retinális és choroideális atrófiát figyeltünk meg. A fundus autofluoreszcencia a károsodásnak megfelelően kifejezetten csökkent volt (7. és 8. ábra).
A genetikai vizsgálat heterozigóta kereteltolódást okozó frameshift variánst, a BBS10 gén c.1024dup, p.(Ile342Asnfs*20) és egy heterozigóta missense variánst, BBS10 c.712G>A, p.(Gly238Ser) igazolt.
Megbeszélés
Mindkét beteg esetében a BBS több szervrendszert érintő általános tünetei mellett súlyos DPR szemészeti képét láthattuk. Az első esetben négy, a másodikban mind a hat elsődleges tünettel rendelkezett a páciens. A genetikai vizsgálat mindkét esetben a Bardet–Biedl-szindróma 10-es altípusát igazolta, 3 esetben missense, míg egy esetben frameshift mutáció fordult elő.
Grudzinska Pechhacker és munkatársai 2021-es közleményükben leírták, hogy a retina degenerációja korábban és súlyosabban jelentkezik a BBS10-es, mint a BBS1-es variánsban szenvedőknél. Tehát valószínű, hogy a látásfunkció progressziója összefügg a BBS genetikai altípusaival (5). Egy másik közleményben olvashatjuk, hogy a BBS2 és a BBS10 génmutációval rendelkező betegeknél gyakoribb a polydactylia és a veseanomáliák előfordulása, mint a BBS1-mutációval rendelkező betegeknél (6).
Feuillan és munkatársai leírták, hogy a BBS-ben szenvedő betegeknél magasabb volt a leptinszint, mint ahogy azt az adipozitásuk mértéke alapján várták, ennek a hátterében leptinrezisztenciát feltételeztek. Továbbá a BBS1-et és BBS10-et összehasonlítva kimutatták, hogy a BBS10-mutációval rendelkező betegeknél szignifikánsan magasabb volt a BMI, nagyobb a zsigeri elhízás és nagyobb az inzulinrezisztencia, mint a BBS1-mutációval rendelkezőknél (3).
Két vizsgált betegünk alapján megerősíthetjük, hogy a BBS10-nél jellemzően rendkívül súlyos formában és korai életkorban jelentkezik a nagyfokú látáscsökkenés és a vesekárosodás, illetve jelentős az elhízás, ami rendszeresen másodlagos kórképekhez is vezet.
Nyilatkozat
A szerzők kijelentik, hogy az esetismertetés megírásával kapcsolatban nem áll fenn velük szemben pénzügyi vagy egyéb lényeges összeütközés, összeférhetetlenségi ok, amely befolyásolhatja a közleményben bemutatott eredményeket, az abból levont következtetéseket vagy azok értelmezését.
Irodalom
1. Baker K, Beales PL. Making sense of cilia in disease: The human ciliopathies. Am J Med Genet C Semin Med Genet 2009; 15; 151(4): 281–295. https://doi.org/10.1002/ajmg.c.30231.
https://doi.org/10.1097/00006982-199515020-00006
2. Dong X, Li Z, Wang D, et al. Prenatal diagnosis of Bardet‑Biedl syndrome due to novel variants in the BBS10 gene in a fetus with multiple anomalies: A case report. Exp Ther Med 2022; 24(6): 721.
https://doi.org/10.3892/etm.2022.11657
3. Feuillan PP, Ng D, Han JC, et al. Patients with Bardet-Biedl syndrome have hyperleptinemia suggestive of leptin resistance. J Clin Endocrinol Metab 2011; 96(3): E528–35.
https://doi.org/10.1210/jc.2010-2290
4. Forsythe E, Beales PL. Bardet-Biedl syndrome. Eur J Hum Genet 2013; 21(1): 8–13.
https://doi.org/10.1038/ejhg.2012.115
5. Grudzinska Pechhacker MK, Jacobson SG, Drack AV, et al. Comparative Natural History of Visual Function from Patients with Biallelic Variants in BBS1 and BBS10. Invest Ophthalmol Vis Sci 2021; 62(15): 26.
https://doi.org/10.1167/iovs.62.15.26
6. Niederlova V, Modrak M, Tsyklauri O, et al. Meta-analysis of genotype-phenotype associations in Bardet-Biedl syndrome uncovers differences among causative genes. Hum Mutat 2019; 40(11): 2068–2087.
https://doi.org/10.1002/humu.23862
7. Suspitsin EN, Imyanitov EN. Bardet-Biedl Syndrome. Mol Syndromol 2016; 7(2): 62–71.
https://doi.org/10.1159/000445491